Letters in Drug Design & Discovery ( IF 1.2 ) Pub Date : 2021-05-01 , DOI: 10.2174/1570180817999201123163617 Vivek Yadav 1 , Rajiv Kumar Tonk 1 , Ramchander Khatri 1
|
Background: ALK inhibitors have become a plausible option for anticancer therapy with the availability of several FDA-approved molecules and clinical trial candidates. Hence, the design of new ALK inhibitors using computational molecular docking studies on the existing inhibitors, is an attractive approach for anticancer drug discovery.
Methods: We generated six types of independent models through structural based molecular docking study, three-dimensional quantitative structure-activity relationship (3D-QSAR) study, and 2DQSAR approaches using different fingerprints, such as dendritic, linear, 2D molprint, and radial.
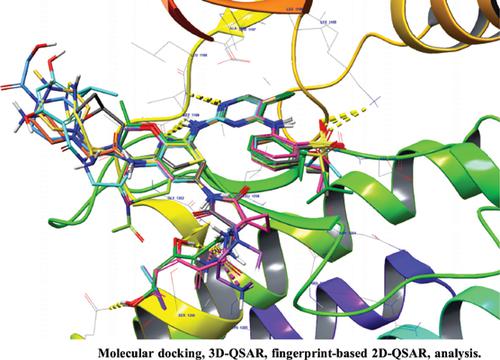
Results: Comparison of the generated models showed that the hinge region hydrogen bond interacted with amino acids ASP1206, MET1199, and LYS1150 in docking analysis and the hydrophobic interacted with amino acids GLU1210, ARG1209, SER1206, and LYS1205 residues are responsible for the ALK inhibition. In the 3D-QSAR study, the hydrogen bond donor features of 2,4- diaryl aminopyrimidine substituents, isopropyl phenyl ring groups in hydrophobic features, and electron-withdrawing groups matched the generated contour plots. The 2D-QSAR fingerprint studies indicated that higher potency was associated with the 2-hydroxy-5-isopropyl benzamide functional group and substituted phenylamine at the second position of the pyrimidine group.
Conclusion: We conclude that the incorporation of these functional groups in the design of new molecules may result in more potent ALK inhibitors.
中文翻译:
分子对接、3D-QSAR、基于指纹的 2D-QSAR、嘧啶分析和 ALK(间变性淋巴瘤激酶)抑制剂的类似物作为抗癌剂
背景:随着几种 FDA 批准的分子和临床试验候选药物的出现,ALK 抑制剂已成为抗癌治疗的一种合理选择。因此,使用现有抑制剂的计算分子对接研究设计新的 ALK 抑制剂是一种有吸引力的抗癌药物发现方法。
方法:我们通过基于结构的分子对接研究、三维定量构效关系 (3D-QSAR) 研究和使用不同指纹的 2DQSAR 方法(如树突、线性、2D molprint 和径向)生成了六种类型的独立模型。
结果:生成的模型的比较表明,在对接分析中铰链区氢键与氨基酸 ASP1206、MET1199 和 LYS1150 相互作用,与氨基酸 GLU1210、ARG1209、SER1206 和 LYS1205 相互作用的疏水性残基负责 ALK 抑制。在 3D-QSAR 研究中,2,4-二芳基氨基嘧啶取代基的氢键供体特征、疏水特征中的异丙基苯环基团和吸电子基团与生成的等高线图相匹配。2D-QSAR 指纹研究表明,更高的效力与 2-羟基-5-异丙基苯甲酰胺官能团和嘧啶基团第二位的取代苯胺有关。
结论:我们得出结论,在新分子的设计中加入这些官能团可能会产生更有效的 ALK 抑制剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号