当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exploration of potential inhibitors for tuberculosis via structure-based drug design, molecular docking, and molecular dynamics simulation studies
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-07-02 , DOI: 10.1002/jcc.26712 Sreerama Rajasekhar 1 , Ramanathan Karuppasamy 2 , Kaushik Chanda 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-07-02 , DOI: 10.1002/jcc.26712 Sreerama Rajasekhar 1 , Ramanathan Karuppasamy 2 , Kaushik Chanda 1
Affiliation
|
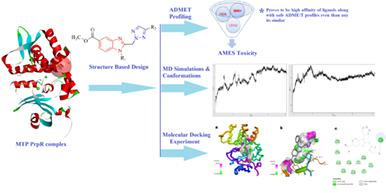
Drug resistance in tuberculosis is major threat to human population. In the present investigation, we aimed to identify novel and potent benzimidazole molecules to overcome the resistance management. A series of 20 benzimidazole derivatives were examined for its activity as selective antitubercular agents. Initially, AutodockVina algorithm was performed to assess the efficacy of the molecules. The results are further enriched by redocking by means of Glide algorithm. The binding free energies of the compounds were then calculated by MM-generalized-born surface area method. Molecular docking studies elucidated that benzimidazole derivatives has revealed formation of hydrogen bond and strong binding affinity in the active site of Mycobacterium tuberculosis protein. Note that ARG308, GLY189, VAL312, LEU403, and LEU190 amino acid residues of Mycobacterium tuberculosis protein PrpR are involved in binding with ligands of benzimidazoles. Interestingly, the ligands exhibited same binding potential to the active site of protein complex PrpR in both the docking programs. In essence, the result portrays that benzimidazole derivatives such as 1p, 1q, and 1 t could be potent and selective antitubercular agents than the standard drug isoniazid. These compounds were then subjected to molecular dynamics simulation to validate the dynamics activity of the compounds against PrpR. Finally, the inhibitory behavior of compounds was predicted using a machine learning algorithm trained on a data collection of 15,000 compounds utilizing graph-based signatures. Overall, the study concludes that designed benzimidazoles can be employed as antitubercular agents. Indeed, the results are helpful for the experimental biologists to develop safe and non-toxic drugs against tuberculosis.
中文翻译:

通过基于结构的药物设计、分子对接和分子动力学模拟研究探索结核病的潜在抑制剂
结核病的耐药性是人类面临的主要威胁。在目前的调查中,我们的目标是确定新的和有效的苯并咪唑分子来克服耐药性管理。检查了一系列 20 种苯并咪唑衍生物作为选择性抗结核剂的活性。最初,执行 AutodockVina 算法来评估分子的功效。通过 Glide 算法重新对接进一步丰富了结果。然后通过MM-广义出生表面积法计算化合物的结合自由能。分子对接研究表明,苯并咪唑衍生物已在结核分枝杆菌的活性部位形成氢键和强结合亲和力蛋白质。请注意,结核分枝杆菌蛋白 PrpR 的ARG308、GLY189、VAL312、LEU403 和 LEU190 氨基酸残基参与与苯并咪唑配体的结合。有趣的是,在两个对接程序中,配体对蛋白质复合物 PrpR 的活性位点表现出相同的结合潜力。从本质上讲,结果表明苯并咪唑衍生物如1p、1q和1 t可能是比标准药物异烟肼更有效和选择性的抗结核药物。然后对这些化合物进行分子动力学模拟,以验证化合物对 PrpR 的动力学活性。最后,使用基于图的特征在 15,000 种化合物的数据集上训练的机器学习算法来预测化合物的抑制行为。总体而言,该研究得出的结论是,设计的苯并咪唑可用作抗结核药物。事实上,这些结果有助于实验生物学家开发安全无毒的抗结核药物。
更新日期:2021-07-23
中文翻译:
通过基于结构的药物设计、分子对接和分子动力学模拟研究探索结核病的潜在抑制剂
结核病的耐药性是人类面临的主要威胁。在目前的调查中,我们的目标是确定新的和有效的苯并咪唑分子来克服耐药性管理。检查了一系列 20 种苯并咪唑衍生物作为选择性抗结核剂的活性。最初,执行 AutodockVina 算法来评估分子的功效。通过 Glide 算法重新对接进一步丰富了结果。然后通过MM-广义出生表面积法计算化合物的结合自由能。分子对接研究表明,苯并咪唑衍生物已在结核分枝杆菌的活性部位形成氢键和强结合亲和力蛋白质。请注意,结核分枝杆菌蛋白 PrpR 的ARG308、GLY189、VAL312、LEU403 和 LEU190 氨基酸残基参与与苯并咪唑配体的结合。有趣的是,在两个对接程序中,配体对蛋白质复合物 PrpR 的活性位点表现出相同的结合潜力。从本质上讲,结果表明苯并咪唑衍生物如1p、1q和1 t可能是比标准药物异烟肼更有效和选择性的抗结核药物。然后对这些化合物进行分子动力学模拟,以验证化合物对 PrpR 的动力学活性。最后,使用基于图的特征在 15,000 种化合物的数据集上训练的机器学习算法来预测化合物的抑制行为。总体而言,该研究得出的结论是,设计的苯并咪唑可用作抗结核药物。事实上,这些结果有助于实验生物学家开发安全无毒的抗结核药物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号