Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2021-06-26 , DOI: 10.1016/j.jmgm.2021.107973 Ayesha Hanif 1 , Rida Kiran 1 , Rasheed Ahmad Khera 1 , Ayesha Ayoub 1 , Khurshid Ayub 2 , Javed Iqbal 3
|
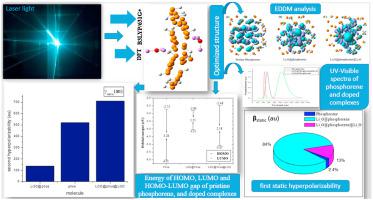
In this study, the nonlinear optical (NLO) properties of pristine phosphorene and superalkali (Li3O) doped phosphorene are estimated through the density functional theory (DFT) method to investigate the optical properties. The geometries of complexes have been optimized using the B3LYP/6-31G (d, p) level of theory. The effects of doping on phosphorene have been thoroughly explained by vertical ionization energy (VIE), interaction energies (Eint), and natural bond orbitals (NBO), Moreover, the density of states (DOS), electron density difference map (EDDM) analysis, the frontier molecular orbitals (FMO) plots are also given out to find more physical divination into the electronic communication and structure property relationship. The doping of superalkali conclusively has reduced the HOMO-LUMO energy gap of M1 3.28 eV–1.25 eV for M2 making it the n-type semiconductor. The higher values of Eint, Efm and VIE obtained for M2 has indicated that this complex has higher stability and stronger interaction between superalkalis and phosphorene. More interestingly, there has been a gradual increase in the first static hyperpolarizability (βstatic) values for M1, M2 and M3 are 115.75 au, 4118.6 au, and 659.30 au respectively. The Static second hyperpolarizability (γstatic) of the doped complexes has also been calculated from which the M2 has the highest value of 1382.5 ҳ 103 au. The TD-DFT exploration has exhibited that the doped molecules are adequately transparent in the UV region. Some selected systems are also compared with the p-NA reference molecule which is a familiar external reference molecule for NLO applications. From UV absorption analysis, it can be found that these doped complexes of phosphorene may be contemplated as a new applicant for intense ultraviolet NLO materials. Computational studies have revealed the stability of M2 and M3 making them feasible as NLO materials in optoelectronic applications.
中文翻译:
调节超碱掺杂磷烯的光电特性
在本研究中,通过密度泛函理论 (DFT) 方法估计原始磷烯和超碱 (Li 3 O) 掺杂磷烯的非线性光学 (NLO) 性质,以研究光学性质。配合物的几何结构已使用 B3LYP/6-31G (d, p) 理论水平进行了优化。掺杂对磷烯的影响已经通过垂直电离能 (VIE)、相互作用能 ( E int), 和自然键轨道 (NBO), 此外, 状态密度 (DOS), 电子密度差图 (EDDM) 分析, 前沿分子轨道 (FMO) 图也被给出,以找到更多的物理占卜进入电子通信和结构属性关系。superalkali的掺杂决定性降低的HOMO-LUMO能隙M1 3.28 EV-1.25电子伏特为M2使其成为n型半导体。M2获得的E int、 E fm和 VIE值较高表明该配合物具有更高的稳定性和强碱与磷烯之间的更强相互作用。更有趣的是,第一次静态超极化(βstatic ) M1、M2和M3 的值分别为115.75 au、4118.6 au 和 659.30 au。还计算了掺杂复合物的静态第二超极化率 (γ static ),从中M2具有最高值 1382.5 ҳ 10 3 au。TD-DFT 探索表明,掺杂分子在 UV 区域是足够透明的。一些选定的系统也与p进行了比较-NA 参考分子,它是 NLO 应用中熟悉的外部参考分子。从紫外吸收分析可以发现,这些掺杂的磷烯复合物可以作为强紫外非线性光学材料的新申请者。计算研究表明M2和M3的稳定性使其在光电应用中作为 NLO 材料是可行的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号