当前位置:
X-MOL 学术
›
Chemistryopen
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Approaches to Discover Novel Natural Compounds for SARS-CoV-2 Therapeutics
ChemistryOpen ( IF 2.5 ) Pub Date : 2021-05-19 , DOI: 10.1002/open.202000332 Shailima Rampogu 1 , Gihwan Lee 1 , Apoorva M Kulkarni 1 , Donghwan Kim 1 , Sanghwa Yoon 1 , Myeong Ok Kim 2 , Keun Woo Lee 1
ChemistryOpen ( IF 2.5 ) Pub Date : 2021-05-19 , DOI: 10.1002/open.202000332 Shailima Rampogu 1 , Gihwan Lee 1 , Apoorva M Kulkarni 1 , Donghwan Kim 1 , Sanghwa Yoon 1 , Myeong Ok Kim 2 , Keun Woo Lee 1
Affiliation
|
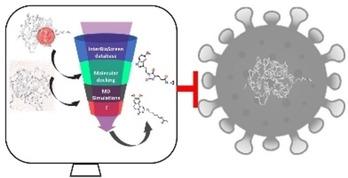
Scientists all over the world are facing a challenging task of finding effective therapeutics for the coronavirus disease (COVID-19). One of the fastest ways of finding putative drug candidates is the use of computational drug discovery approaches. The purpose of the current study is to retrieve natural compounds that have obeyed to drug-like properties as potential inhibitors. Computational molecular modelling techniques were employed to discover compounds with potential SARS-CoV-2 inhibition properties. Accordingly, the InterBioScreen (IBS) database was obtained and was prepared by minimizing the compounds. To the resultant compounds, the absorption, distribution, metabolism, excretion and toxicity (ADMET) and Lipinski's Rule of Five was applied to yield drug-like compounds. The obtained compounds were subjected to molecular dynamics simulation studies to evaluate their stabilities. In the current article, we have employed the docking based virtual screening method using InterBioScreen (IBS) natural compound database yielding two compounds has potential hits. These compounds have demonstrated higher binding affinity scores than the reference compound together with good pharmacokinetic properties. Additionally, the identified hits have displayed stable interaction results inferred by molecular dynamics simulation results. Taken together, we advocate the use of two natural compounds, STOCK1N-71493 and STOCK1N-45683 as SARS-CoV-2 treatment regime.
中文翻译:

发现用于 SARS-CoV-2 治疗的新型天然化合物的计算方法
世界各地的科学家都面临着寻找冠状病毒病(COVID-19)有效疗法的艰巨任务。寻找假定候选药物的最快方法之一是使用计算药物发现方法。当前研究的目的是检索具有类似药物特性的天然化合物作为潜在的抑制剂。采用计算分子建模技术来发现具有潜在 SARS-CoV-2 抑制特性的化合物。因此,获得了 InterBioScreen (IBS) 数据库,并通过最小化化合物来制备。对于所得化合物,应用吸收、分布、代谢、排泄和毒性(ADMET)和利平斯基五法则来产生药物样化合物。对所得化合物进行分子动力学模拟研究以评估其稳定性。在本文中,我们采用基于对接的虚拟筛选方法,使用 InterBioScreen (IBS) 天然化合物数据库产生两种具有潜在命中的化合物。这些化合物已表现出比参考化合物更高的结合亲和力分数以及良好的药代动力学特性。此外,通过分子动力学模拟结果推断,所识别的命中显示出稳定的相互作用结果。综上所述,我们主张使用两种天然化合物STOCK1N-71493和STOCK1N-45683作为 SARS-CoV-2 的治疗方案。
更新日期:2021-05-20
中文翻译:
发现用于 SARS-CoV-2 治疗的新型天然化合物的计算方法
世界各地的科学家都面临着寻找冠状病毒病(COVID-19)有效疗法的艰巨任务。寻找假定候选药物的最快方法之一是使用计算药物发现方法。当前研究的目的是检索具有类似药物特性的天然化合物作为潜在的抑制剂。采用计算分子建模技术来发现具有潜在 SARS-CoV-2 抑制特性的化合物。因此,获得了 InterBioScreen (IBS) 数据库,并通过最小化化合物来制备。对于所得化合物,应用吸收、分布、代谢、排泄和毒性(ADMET)和利平斯基五法则来产生药物样化合物。对所得化合物进行分子动力学模拟研究以评估其稳定性。在本文中,我们采用基于对接的虚拟筛选方法,使用 InterBioScreen (IBS) 天然化合物数据库产生两种具有潜在命中的化合物。这些化合物已表现出比参考化合物更高的结合亲和力分数以及良好的药代动力学特性。此外,通过分子动力学模拟结果推断,所识别的命中显示出稳定的相互作用结果。综上所述,我们主张使用两种天然化合物STOCK1N-71493和STOCK1N-45683作为 SARS-CoV-2 的治疗方案。









































 京公网安备 11010802027423号
京公网安备 11010802027423号