Current Bioinformatics ( IF 2.4 ) Pub Date : 2021-02-28 , DOI: 10.2174/1574893615999200707141926 Affan Alim 1 , Abdul Rafay 1 , Imran Naseem 2
|
Background: Proteins contribute significantly in every task of cellular life. Their functions encompass the building and repairing of tissues in human bodies and other organisms. Hence they are the building blocks of bones, muscles, cartilage, skin, and blood. Similarly, antifreeze proteins are of prime significance for organisms that live in very cold areas. With the help of these proteins, the cold water organisms can survive below zero temperature and resist the water crystallization process, which may cause the rupture in the internal cells and tissues. AFP’s have also attracted attention and interest in food industries and cryopreservation.
Objective: With the increase in the availability of genomic sequence data of protein, an automated and sophisticated tool for AFP recognition and identification is in dire need. The sequence and structures of AFP are highly distinct, therefore, most of the proposed methods fail to show promising results on different structures. A consolidated method is proposed to produce a competitive performance on a highly distinct AFP structure.
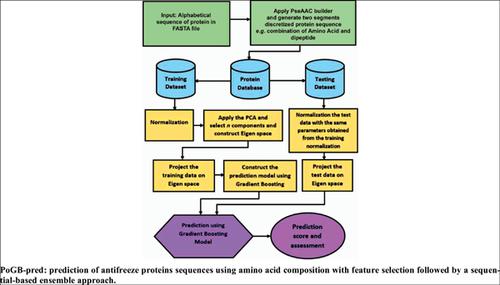
Methods: In this study, machine learning-based algorithms including Principal Component Analysis (PCA) followed by Gradient Boosting (GB) were proposed to be used for anti-freeze protein identification. To analyze the performance and validation of the proposed model, various combinations of two segments' composition of amino acid and dipeptides are used. PCA, in particular, is proposed for dimension reduction and high variance retaining of data, which is followed by an ensemble method named gradient boosting for modeling and classification.
Results: The proposed method obtained a superfluous performance on PDB, Pfam, and Uniprot datasets as compared to the RAFP-Pred method. In experiment-3, by utilizing only 150 PCA components, a high accuracy of 89.63% was achieved, which is superior to 87.41% utilizing 300 significant features reported for the RAFP-Pred method. Experiment-2 is conducted using two different datasets such that non-AFP from the PISCES server and AFPs from Protein data bank. In this experiment-2, the proposed method attained high sensitivity of 79.16% which is 12.50% better than state-of-the-art RAFP-pred method.
Conclusion: AFPs have a common function with a distinct structure. Therefore, the development of a single model for different sequences often fails for AFPs. Robust results have been shown by the proposed model on the diversity of training and testing datasets. The results of the proposed model outperformed compared to the previous AFPs prediction method, such as RAFP-Pred. The proposed model consists of PCA for dimension reduction, followed by gradient boosting for classification. Due to simplicity, scalability properties, and high performance result, this model can be easily extended for analyzing the proteomic and genomic datasets.
中文翻译:
PoGB-pred:使用具有特征选择的氨基酸组成,然后基于序列的集成方法来预测抗冻蛋白序列
背景:蛋白质在细胞生命的每个任务中均起着重要作用。它们的功能包括构建和修复人体和其他生物体中的组织。因此,它们是骨骼,肌肉,软骨,皮肤和血液的基础。同样,抗冻蛋白对于生活在非常寒冷地区的生物也具有重要意义。借助这些蛋白质,冷水生物可以在零温度以下生存并抵抗水的结晶过程,这可能会导致内部细胞和组织破裂。法新社也引起了人们对食品工业和冷冻保存的关注。
目的:随着蛋白质基因组序列数据可用性的增加,迫切需要一种用于AFP识别和鉴定的自动化且复杂的工具。AFP的序列和结构是高度不同的,因此,大多数提出的方法未能在不同的结构上显示出令人鼓舞的结果。提出了一种合并方法,以在高度不同的AFP结构上产生竞争绩效。
方法:在这项研究中,基于机器学习的算法(包括主成分分析(PCA)和梯度增强(GB))被建议用于抗冻蛋白鉴定。为了分析所提出模型的性能和验证,使用了氨基酸和二肽两个片段组成的各种组合。尤其是,提出了PCA用于数据的降维和高方差保留,随后提出了一种称为梯度提升的集成方法,用于建模和分类。
结果:与RAFP-Pred方法相比,该方法在PDB,Pfam和Uniprot数据集上获得了多余的性能。在实验3中,通过仅使用150个PCA组件,获得了89.63%的高精度,这优于使用RAFP-Pred方法报道的300个重要特征的87.41%的精度。实验2是使用两个不同的数据集进行的,例如来自PISCES服务器的非AFP和来自蛋白质数据库的AFP。在本实验2中,拟议的方法获得了79.16%的高灵敏度,比最先进的RAFP-pred方法高出12.50%。
结论:AFP具有共同的功能,结构独特。因此,对于AFP,针对不同序列开发单个模型通常会失败。所提出的模型在训练和测试数据集的多样性上已显示出稳健的结果。与先前的AFP预测方法(如RAFP-Pred)相比,所提出模型的结果表现出色。所提出的模型包括PCA用于降维,然后是梯度增强用于分类。由于具有简单性,可伸缩性和高性能结果,可以轻松扩展此模型以分析蛋白质组学和基因组数据集。










































 京公网安备 11010802027423号
京公网安备 11010802027423号