当前位置:
X-MOL 学术
›
J. Neuroendocrinol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome
Journal of Neuroendocrinology ( IF 3.3 ) Pub Date : 2021-05-18 , DOI: 10.1111/jne.12994 Felipe Correa-da-Silva 1, 2, 3 , Eric Fliers 1 , Dick F Swaab 3 , Chun-Xia Yi 1, 2, 3
Journal of Neuroendocrinology ( IF 3.3 ) Pub Date : 2021-05-18 , DOI: 10.1111/jne.12994 Felipe Correa-da-Silva 1, 2, 3 , Eric Fliers 1 , Dick F Swaab 3 , Chun-Xia Yi 1, 2, 3
Affiliation
|
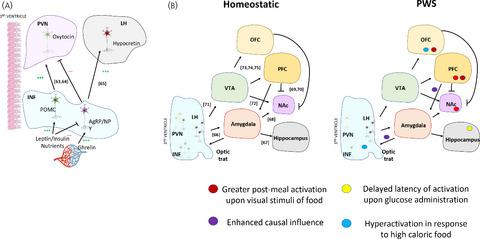
Prader-Willi Syndrome (PWS) is a rare and incurable congenital neurodevelopmental disorder, resulting from the absence of expression of a group of genes on the paternally acquired chromosome 15q11-q13. Phenotypical characteristics of PWS include infantile hypotonia, short stature, incomplete pubertal development, hyperphagia and morbid obesity. Hypothalamic dysfunction in controlling body weight and food intake is a hallmark of PWS. Neuroimaging studies have demonstrated that PWS subjects have abnormal neurocircuitry engaged in the hedonic and physiological control of feeding behavior. This is translated into diminished production of hypothalamic effector peptides which are responsible for the coordination of energy homeostasis and satiety. So far, studies with animal models for PWS and with human post-mortem hypothalamic specimens demonstrated changes particularly in the infundibular and the paraventricular nuclei of the hypothalamus, both in orexigenic and anorexigenic neural populations. Moreover, many PWS patients have a severe endocrine dysfunction, e.g. central hypogonadism and/or growth hormone deficiency, which may contribute to the development of increased fat mass, especially if left untreated. Additionally, the role of non-neuronal cells, such as astrocytes and microglia in the hypothalamic dysregulation in PWS is yet to be determined. Notably, microglial activation is persistently present in non-genetic obesity. To what extent microglia, and other glial cells, are affected in PWS is poorly understood. The elucidation of the hypothalamic dysfunction in PWS could prove to be a key feature of rational therapeutic management in this syndrome. This review aims to examine the evidence for hypothalamic dysfunction, both at the neuropeptidergic and circuitry levels, and its correlation with the pathophysiology of PWS.
中文翻译:

Prader Willi 综合征中的下丘脑神经肽和神经电路
Prader-Willi 综合征 (PWS) 是一种罕见且无法治愈的先天性神经发育障碍,由父系获得的染色体 15q11-q13 上的一组基因缺乏表达所致。PWS 的表型特征包括婴儿肌张力减退、身材矮小、青春期发育不完全、食欲过盛和病态肥胖。控制体重和食物摄入的下丘脑功能障碍是 PWS 的标志。神经影像学研究表明,PWS 受试者具有异常的神经回路,参与进食行为的享乐和生理控制。这转化为负责协调能量稳态和饱腹感的下丘脑效应肽的产生减少。迄今为止,对 PWS 的动物模型和人类死后下丘脑标本的研究表明,特别是下丘脑漏斗核和室旁核的变化,在食欲和厌食性神经群中都发生了变化。此外,许多 PWS 患者有严重的内分泌功能障碍,例如中枢性腺功能减退症和/或生长激素缺乏症,这可能导致脂肪量增加,尤其是如果不治疗的话。此外,非神经元细胞,如星形胶质细胞和小胶质细胞在 PWS 下丘脑失调中的作用尚待确定。值得注意的是,小胶质细胞激活持续存在于非遗传性肥胖中。PWS 对小胶质细胞和其他神经胶质细胞的影响程度尚不清楚。PWS 下丘脑功能障碍的阐明可能被证明是该综合征合理治疗管理的一个关键特征。本综述旨在检查下丘脑功能障碍的证据,包括神经肽能和电路水平,及其与 PWS 病理生理学的相关性。
更新日期:2021-07-26
中文翻译:
Prader Willi 综合征中的下丘脑神经肽和神经电路
Prader-Willi 综合征 (PWS) 是一种罕见且无法治愈的先天性神经发育障碍,由父系获得的染色体 15q11-q13 上的一组基因缺乏表达所致。PWS 的表型特征包括婴儿肌张力减退、身材矮小、青春期发育不完全、食欲过盛和病态肥胖。控制体重和食物摄入的下丘脑功能障碍是 PWS 的标志。神经影像学研究表明,PWS 受试者具有异常的神经回路,参与进食行为的享乐和生理控制。这转化为负责协调能量稳态和饱腹感的下丘脑效应肽的产生减少。迄今为止,对 PWS 的动物模型和人类死后下丘脑标本的研究表明,特别是下丘脑漏斗核和室旁核的变化,在食欲和厌食性神经群中都发生了变化。此外,许多 PWS 患者有严重的内分泌功能障碍,例如中枢性腺功能减退症和/或生长激素缺乏症,这可能导致脂肪量增加,尤其是如果不治疗的话。此外,非神经元细胞,如星形胶质细胞和小胶质细胞在 PWS 下丘脑失调中的作用尚待确定。值得注意的是,小胶质细胞激活持续存在于非遗传性肥胖中。PWS 对小胶质细胞和其他神经胶质细胞的影响程度尚不清楚。PWS 下丘脑功能障碍的阐明可能被证明是该综合征合理治疗管理的一个关键特征。本综述旨在检查下丘脑功能障碍的证据,包括神经肽能和电路水平,及其与 PWS 病理生理学的相关性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号