Comparative Biochemistry and Physiology B: Biochemistry & Molecular Biology ( IF 2.2 ) Pub Date : 2021-05-15 , DOI: 10.1016/j.cbpb.2021.110616 Valentina Lukinović 1 , Kyle K Biggar 1
|
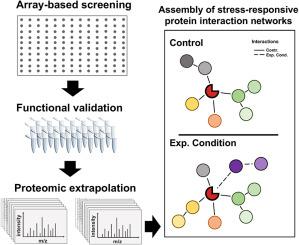
Following the decoding of the first human genome, researchers have vastly improved their understanding of cell biology and its regulation. As a result, it has become clear that it is not merely genetic information, but the aberrant changes in the functionality and connectivity of its encoded proteins that drive cell response to periods of stress and external cues. Therefore, proper utilization of refined methods that help to describe protein signalling or regulatory networks (i.e., functional connectivity), can help us understand how change in the signalling landscape effects the cell. However, given the vast complexity in ‘how and when’ proteins communicate or interact with each other, it is extremely difficult to define, characterize, and understand these interaction networks in a tangible manner. Herein lies the challenge of tackling the functional proteome; its regulation is encoded in multiple layers of interaction, chemical modification and cell compartmentalization. To address and refine simple research questions, modern reductionist strategies in protein biochemistry have successfully used peptide-based experiments; their summation helping to simplify the overall complexity of these protein interaction networks. In this way, peptides are powerful tools used in fundamental research that can be readily applied to comparative biochemical research. Understanding and defining how proteins interact is one of the key aspects towards understanding how the proteome functions. To date, reductionist peptide-based research has helped to address a wide range of proteome-related research questions, including the prediction of enzymes substrates, identification of posttranslational modifications, and the annotation of protein interaction partners. Peptide arrays have been used to identify the binding specificity of reader domains, which are able to recognise the posttranslational modifications; forming dynamic protein interactions that are dependent on modification state. Finally, representing one of the fastest growing classes of inhibitor molecules, peptides are now begin explored as “disruptors” of protein-protein interactions or enzyme activity. Collectively, this review will discuss the use of peptides, peptide arrays, peptide-oriented computational biochemistry as modern reductionist strategies in deconvoluting the functional proteome.
中文翻译:
通过肽生物化学中的还原策略解卷积复杂的蛋白质相互作用网络:现代方法和研究问题
在对第一个人类基因组进行解码后,研究人员极大地提高了他们对细胞生物学及其调控的理解。因此,很明显,它不仅是遗传信息,而且是其编码蛋白质的功能和连通性的异常变化,驱动细胞对压力和外部线索的时期做出反应。因此,适当利用有助于描述蛋白质信号传导或调控网络的精细方法(即,功能连接),可以帮助我们了解信号景观的变化如何影响细胞。然而,考虑到蛋白质“如何以及何时”相互交流或相互作用的巨大复杂性,以有形的方式定义、表征和理解这些相互作用网络是极其困难的。这就是解决功能蛋白质组的挑战;它的调节被编码在多层相互作用、化学修饰和细胞区室化中。为了解决和完善简单的研究问题,蛋白质生物化学中的现代还原策略已成功使用基于肽的实验;它们的总和有助于简化这些蛋白质相互作用网络的整体复杂性。通过这种方式,肽是基础研究中使用的强大工具,可以很容易地应用于比较生化研究。理解和定义蛋白质如何相互作用是理解蛋白质组功能的关键方面之一。迄今为止,基于还原论的肽研究已经帮助解决了广泛的蛋白质组相关研究问题,包括酶底物的预测、翻译后修饰的识别以及蛋白质相互作用伙伴的注释。肽阵列已被用于识别阅读器域的结合特异性,能够识别翻译后修饰;形成依赖于修饰状态的动态蛋白质相互作用。最后,代表了增长最快的一类抑制剂分子,现在开始探索肽作为蛋白质-蛋白质相互作用或酶活性的“破坏者”。总的来说,这篇综述将讨论使用肽、肽阵列、面向肽的计算生物化学作为现代还原策略在功能蛋白质组解卷积中的应用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号