Solid State Sciences ( IF 3.4 ) Pub Date : 2021-05-10 , DOI: 10.1016/j.solidstatesciences.2021.106633 Adesh Rohan Mishra , Snehanshu Pal
|
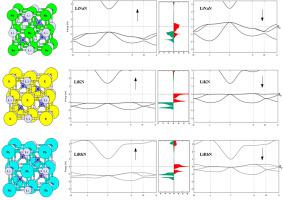
Density functional theory (DFT) incorporating GGA-PBE approximation has been implemented to study structural, electronic, magnetic, mechanical and thermodynamic properties of the d0 Half-Heusler LiXN (X = Na, K, Rb) alloys. According to the spin-polarized calculations, all the LiXN alloys crystallize in the α-phase ferromagnetic ground state configuration. All three LiXN alloys show a total magnetic moment of 1.00 μB. The integer values of the total magnetic moments, along with the spin-polarized electronic band structures and the density of states plots indicate that the LiXN alloys are true half-metals in nature. The half-metallic nature of the LiXN alloys is further confirmed as the total magnetic moments are in complete agreement with the Slater-Pauling rule of 8. The origin of the half-metallic ferromagnetic behaviour has been investigated and it has been observed that the 2p states of N are the major contributors in case of all the three alloys. The alloys retain the characteristic half-metallicity over a wide range of lattice parameters, thus confirming their robustness and usability in a variety of spintronic applications. The mechanical properties of the LiXN alloys have been investigated and it has been observed that LiKN and LiRbN are found to be mechanically unstable, whereas LiNaN is chemically, energetically and mechanically stable. Various thermodynamic properties have been further computed and analysed using the quasi-harmonic approximation (QHA). Specific heat capacity at room temperature (300K), the Debye temperature, and the zero-point energy for the LiNaN alloy as well as the average velocity of sound inside the LiNaN alloy have been evaluated.
中文翻译:
用于研究d 0半霍斯勒LiXN(X = Na,K,Rb)合金的电子结构以及磁,机械和热力学性质的第一性原理计算
已采用结合GGA-PBE近似的密度泛函理论(DFT)来研究d 0的结构,电子,磁性,机械和热力学性质Half-Heusler LiXN(X = Na,K,Rb)合金。根据自旋极化计算,所有的LiXN合金都以α相铁磁基态配置结晶。所有三种LiXN合金的总磁矩均为1.00μB。总磁矩的整数值,以及自旋极化的电子带结构和状态密度图表明,LiXN合金本质上是真正的半金属。LiXN合金的半金属性质得到了进一步证实,因为总磁矩与8的Slater-Pauling规则完全一致。已经研究了半金属铁磁行为的起源,并且观察到2p在这三种合金中,N的状态是主要的贡献者。该合金在很宽的晶格参数范围内都具有特征性的半金属性,因此证实了它们在各种自旋电子学应用中的坚固性和可用性。已经研究了LiXN合金的机械性能,并且已经发现发现LiKN和LiRbN在机械上是不稳定的,而LiNaN在化学上,能量上和机械上都是稳定的。使用准谐波近似(QHA)进一步计算和分析了各种热力学性质。对LiNaN合金在室温(300K)的比热容,德拜温度和零点能量以及LiNaN合金内部的平均声速进行了评估。已经研究了LiXN合金的机械性能,并且已经发现发现LiKN和LiRbN在机械上是不稳定的,而LiNaN在化学上,能量上和机械上都是稳定的。使用准谐波近似(QHA)进一步计算和分析了各种热力学性质。对LiNaN合金在室温(300K)的比热容,德拜温度和零点能量以及LiNaN合金内部的平均声速进行了评估。已经研究了LiXN合金的机械性能,并且已经发现发现LiKN和LiRbN在机械上是不稳定的,而LiNaN在化学上,能量上和机械上都是稳定的。使用准谐波近似(QHA)进一步计算和分析了各种热力学性质。对LiNaN合金在室温(300K)的比热容,德拜温度和零点能量以及LiNaN合金内部的平均声速进行了评估。










































 京公网安备 11010802027423号
京公网安备 11010802027423号