Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2021-05-10 , DOI: 10.1016/j.jmgm.2021.107931 Dean Sherry 1 , Roland Worth 1 , Zaahida Sheik Ismail 1 , Yasien Sayed 1
|
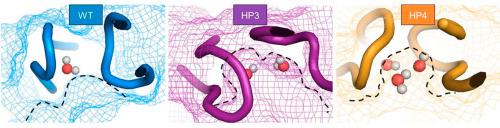
The HIV-1 protease is an important drug target in antiretroviral therapy due to the crucial role it plays in viral maturation. A greater understanding of the dynamics of the protease as a result of drug-induced mutations has been successfully elucidated using computational models in the past. We performed induced-fit docking studies and molecular dynamics simulations on the wild-type South African HIV-1 subtype C protease and two non-active site mutation-containing protease variants; HP3 PR and HP4 PR. The HP3 PR contained the I13V, I62V, and V77I mutations while HP4 PR contained the same mutations with the addition of the L33F mutation. The simulations were initiated in a cubic cell universe containing explicit solvent, with the protease variants beginning in the fully closed conformation. The trajectory for each simulation totalled 50 ns. The results indicate that the mutations increase the dynamics of the flap, hinge, fulcrum and cantilever regions when compared to the wild-type protease while in complex with protease inhibitors. Specifically, these mutations result in the protease favouring the semi-open conformation when in complex with inhibitors. Moreover, the HP4 PR adopted curled flap tip conformers which coordinated several water molecules into the active site in a manner that may reduce inhibitor binding affinity. The mutations affected the thermodynamic landscape of inhibitor binding as there were fewer observable chemical contacts between the mutated variants and saquinavir, atazanavir and darunavir. These data help to elucidate the biophysical basis for the selection of cooperative non-active site mutations by the HI virus.
中文翻译:
HIV蛋白酶中协同非活性位点突变的悬臂为中心的机制:对皮瓣动力学的影响。
由于HIV-1蛋白酶在病毒成熟中起着至关重要的作用,因此它是抗逆转录病毒疗法中的重要药物靶标。过去已经使用计算模型成功阐明了对药物诱导的突变导致的蛋白酶动力学的更深入的了解。我们对野生型南非HIV-1亚型C蛋白酶和两个含有非活性位点突变的蛋白酶变体进行了诱导拟合对接研究和分子动力学模拟。HP3 PR和HP4 PR。HP3 PR包含I13V,I62V和V77I突变,而HP4 PR包含相同的突变以及L33F突变。在含有明确溶剂的立方细胞环境中开始模拟,蛋白酶变体从完全封闭的构象开始。每次模拟的轨迹总计为50 ns。结果表明,与野生型蛋白酶相比,与蛋白酶抑制剂复合时,突变增加了皮瓣,铰链,支点和悬臂区域的动力学。特别地,当与抑制剂复合时,这些突变导致蛋白酶有利于半开放构象。此外,HP4 PR采用了卷曲的襟翼末端构象物,该构象物以减少抑制剂结合亲和力的方式将几个水分子配位到活性位点。突变影响抑制剂结合的热力学格局,因为突变的变体与沙奎那韦,阿扎那韦和达那那韦之间的可观察到的化学接触较少。这些数据有助于阐明HI病毒选择协同非活性位点突变的生物物理基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号