Progress in Neurobiology ( IF 6.7 ) Pub Date : 2021-05-02 , DOI: 10.1016/j.pneurobio.2021.102070 Soumitra Ghosh 1 , Seok Joon Won 1 , Jiejie Wang 1 , Rebecca Fong 1 , Nicholas J M Butler 1 , Arianna Moss 1 , Candance Wong 1 , June Pan 1 , Jennifer Sanchez 1 , Annie Huynh 1 , Long Wu 1 , Fredric P Manfredsson 2 , Raymond A Swanson 1
|
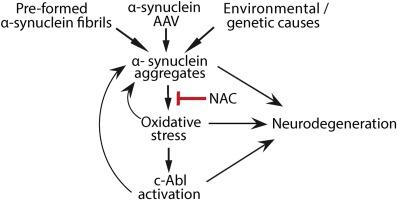
Oxidative stress and α-synuclein aggregation both drive neurodegeneration in Parkinson’s disease, and the protein kinase c-Abl provides a potential amplifying link between these pathogenic factors. Suppressing interactions between these factors may thus be a viable therapeutic approach for this disorder. To evaluate this possibility, pre-formed α-synuclein fibrils (PFFs) were used to induce α-synuclein aggregation in neuronal cultures. Exposure to PFFs induced oxidative stress and c-Abl activation in wild-type neurons. By contrast, α-synuclein - deficient neurons, which cannot form α-synuclein aggregates, failed to exhibit either oxidative stress or c-Abl activation. N-acetyl cysteine, a thiol repletion agent that supports neuronal glutathione metabolism, suppressed the PFF - induced redox stress and c-Abl activation in the wild-type neurons, and likewise suppressed α-synuclein aggregation. Parallel findings were observed in mouse brain: PFF-induced α-synuclein aggregation in the substantia nigra was associated with redox stress, c-Abl activation, and dopaminergic neuronal loss, along with microglial activation and motor impairment, all of which were attenuated with oral N-acetyl cysteine. Similar results were obtained using AAV-mediated α-synuclein overexpression as an alternative means of driving α-synuclein aggregation in vivo. These findings show that α-synuclein aggregates induce c-Abl activation by a redox stress mechanism. c-Abl activation in turn promotes α-synuclein aggregation, in a feed-forward interaction. The capacity of N-acetyl cysteine to interrupt this interaction adds mechanistic support its consideration as a therapeutic in Parkinson’s disease.
中文翻译:
α-突触核蛋白聚集体通过前馈氧化还原应激机制诱导 c-Abl 活化和多巴胺能神经元损失
氧化应激和 α-突触核蛋白聚集都驱动帕金森病的神经变性,蛋白激酶 c-Abl 提供了这些致病因素之间的潜在放大联系。因此,抑制这些因素之间的相互作用可能是治疗这种疾病的一种可行的治疗方法。为了评估这种可能性,使用预先形成的 α-突触核蛋白原纤维 (PFF) 在神经元培养物中诱导 α-突触核蛋白聚集。暴露于 PFFs 在野生型神经元中诱导氧化应激和 c-Abl 激活。相比之下,不能形成 α-突触核蛋白聚集体的 α-突触核蛋白缺陷神经元未能表现出氧化应激或 c-Abl 激活。N-乙酰半胱氨酸是一种硫醇补充剂,支持神经元谷胱甘肽代谢,抑制野生型神经元中 PFF 诱导的氧化还原应激和 c-Abl 活化,并且同样抑制了α-突触核蛋白聚集。在小鼠大脑中观察到了类似的发现:PFF 诱导的黑质中的 α-突触核蛋白聚集与氧化还原应激、c-Abl 激活和多巴胺能神经元丢失以及小胶质细胞激活和运动障碍有关,所有这些都通过口服而减弱N-乙酰半胱氨酸。使用 AAV 介导的 α-突触核蛋白过表达作为驱动 α-突触核蛋白聚集的替代手段获得了类似的结果在体内。这些发现表明,α-突触核蛋白聚集体通过氧化还原应激机制诱导 c-Abl 活化。c-Abl 激活反过来促进 α-突触核蛋白聚集,在前馈相互作用中。N-乙酰半胱氨酸中断这种相互作用的能力增加了将其作为帕金森病治疗剂的机制支持。









































 京公网安备 11010802027423号
京公网安备 11010802027423号