The European Physical Journal B ( IF 1.6 ) Pub Date : 2021-04-27 , DOI: 10.1140/epjb/s10051-021-00090-2 Y. Medkour , F. Djeghloul , N. Bouarissa , A. Roumili
|
Abstract
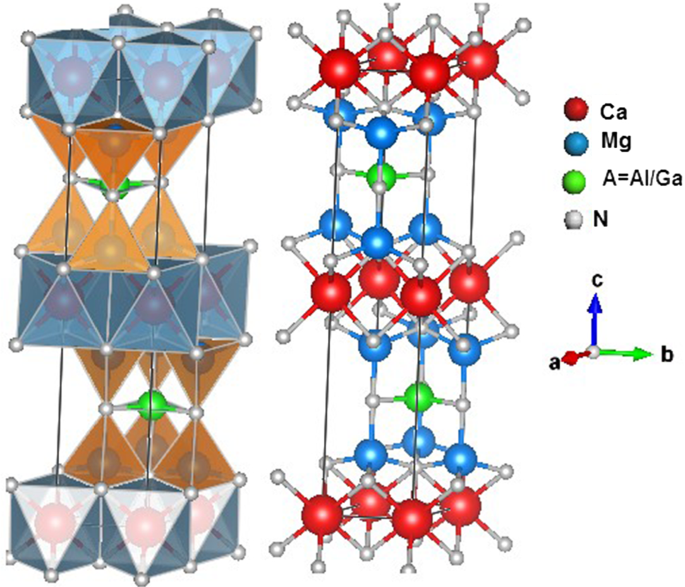
The present paper aims to investigate the structural, elastic, thermodynamic, electronic, and optical properties of \(\hbox {CaMg}_{2}\hbox {AN}_{3}\)(\(\hbox {A}= \hbox {Al}\) and Ga) using first-principle calculations and quasi-harmonic Debye model. The obtained ground-state lattice parameters were in good agreement with the experimental values. Pressure effect on the structural parameters was tested up to 20 GPa, the results reveal that the contractions are higher along the c-axis than along the a-axis. The computed single-crystal elastic moduli show that the unidirectional constant \(\hbox {C}_{11}\) is about 60% greater than \(\hbox {C}_{33}\). Cauchy pressure and Poisson ratio suggest that the chemical bonding in \(\hbox {CaMg}_{2}\hbox {AN}_{3}\) is a mixture of covalent and ionic characters. Elastic anisotropy was discussed using different approaches, and the results show a weak elastic anisotropy. By means of Gibbs program, we have evaluated the thermodynamic properties such as Debye temperature \(\uptheta _{{D}}\), heat capacities \({C}_{{v}}\) and \({C}_{{p}}\), and expansion thermal coefficient under pressure ranging from 0 to 20 GPa and at temperature ranging from 0 to 1500 K for both compounds. The evaluated value of Dulong–Petit limit of both the semiconductors is \(360.5\ \hbox {Jmol}^{-1}\hbox {K}^{-1}\). Band structure curves show a direct band gap of about 1.88 and 0.78 eV for \(\hbox {CaMg}_{2}\hbox {AlN}_{3}\) and \(\hbox {CaMg}_{2} \hbox {GaN}_{3}\), respectively. Density of states and charge densities analysis confirm the predicted ionic-covalent bonding in both nitrides. Additionally, optical functions such as the refractive index, the reflectivity, and the absorption coefficient were calculated and discussed for two polarized radiations.
Graphic abstract
中文翻译:
$$ \ hbox {CaMg} _ {2} \ hbox {AN} _ {3} $$ CaMg 2 AN 3($$ \ hbox {A} = \ hbox { Al} $$ A = Al和Ga):从头算起
摘要
本文旨在研究\(\ hbox {CaMg} _ {2} \ hbox {AN} _ {3} \)(\(\ hbox {A} = \ hbox {Al} \)和Ga),使用第一性原理计算和准谐波Debye模型。获得的基态晶格参数与实验值吻合良好。测试了高达20 GPa的压力对结构参数的影响,结果表明,沿c轴的收缩比沿a轴的收缩高。计算出的单晶弹性模量表明,单向常数\(\ hbox {C} _ {11} \)比\(\ hbox {C} _ {33} \)大约60%。柯西压力和泊松比暗示了化学键的存在。\(\ hbox {CaMg} _ {2} \ hbox {AN} _ {3} \)是共价和离子字符的混合。使用不同的方法讨论了弹性各向异性,结果显示出较弱的弹性各向异性。通过吉布斯程序,我们评估了热力学性质,例如德拜温度\(\ uptheta _ {{D}} \),热容\({C} _ {{v}} \)和\({C} _ {{{p}} \)和两种化合物在0至20 GPa的压力和0至1500 K的温度下的膨胀热系数。两种半导体的Dulong–Petit极限的评估值为\(360.5 \ \ hbox {Jmol} ^ {-1} \ hbox {K} ^ {-1} \)。能带结构曲线显示,直接带隙约为1.88 eV和0.78 eV。\(\ hbox {CaMg} _ {2} \ hbox {AlN} _ {3} \)和\(\ hbox {CaMg} _ {2} \ hbox {GaN} _ {3} \)。状态密度和电荷密度分析证实了两种氮化物中的预测离子共价键。此外,还针对两个偏振辐射计算并讨论了诸如折射率,反射率和吸收系数之类的光学功能。










































 京公网安备 11010802027423号
京公网安备 11010802027423号