Surface Science ( IF 2.1 ) Pub Date : 2021-04-03 , DOI: 10.1016/j.susc.2021.121849 Andréia Luísa da Rosa , Erika Nascimento Lima , Maurício Chagas da Silva , Renato Borges Pontes , Tomé Mauro Schmidt , Thomas Frauenheim
|
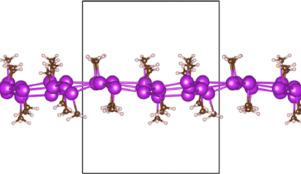
We perform first-principles calculations of the electronic and dielectric properties of bismuthene functionalized with small ligands. Molecular dynamics simulations show that adsorption of these ligands are stable at 300K. We show by calculating the Z topological invariant that all functionalized structures have topological insulating (TI) behavior with a sizable energy band gap. All functional groups induce an electronic change enough to drastically modify the structural and electronic structure of bismuthene. Furthermore, our findings indicate that the dielectric properties exhibit a large anisotropy with the main absorption peak in the visible region.
中文翻译:
通过分子吸附调节铋单层的电子和光学性质
我们执行用小配体官能化的铋的电子和介电性能的第一性原理计算。分子动力学模拟表明,这些配体的吸附在300K时稳定。我们通过计算Z来显示拓扑不变性,即所有功能化结构都具有拓扑绝缘(TI)行为,并且能带隙较大。所有官能团均会引起电子变化,足以彻底改变铋的结构和电子结构。此外,我们的发现表明介电性能表现出较大的各向异性,主要吸收峰位于可见光区域。










































 京公网安备 11010802027423号
京公网安备 11010802027423号