The European Physical Journal D ( IF 1.5 ) Pub Date : 2021-03-19 , DOI: 10.1140/epjd/s10053-021-00120-9 Yu-Guo Xu , Zhao-Jun Han , Jing-Bo Sun , Jie Zhao , Jian-Gang Yao , Deng-Feng Yin
|
Abstract
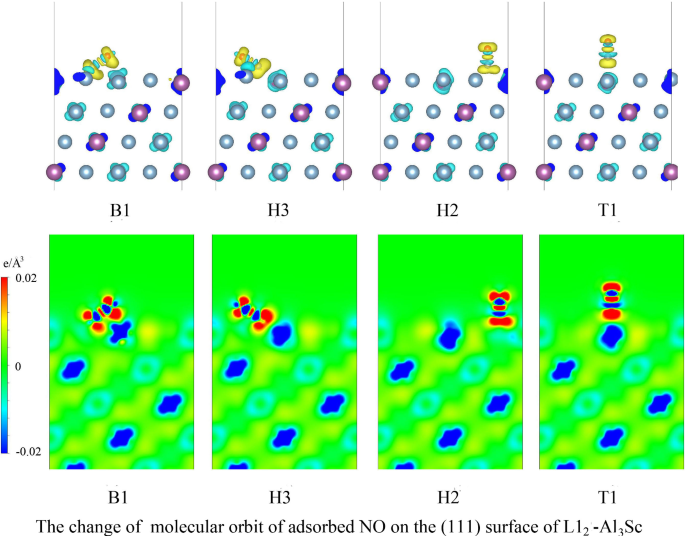
Al-based intermetallic L1\(_{\mathrm {2}}\)-Al\(_{\mathrm {3}}\)Sc exhibits a good performance in adsorbing NO molecule, and the maximum of adsorption energy reaches nearly 3 eV which corresponds to the significant elongation of N–O distance (more than 20% ). The predicted results shows that at the 0.25 coverage, the energetically favorable adsorption configurations for the Al\(_{\mathrm {3}}\)Sc (1 1 1) surface are the bridge and certain hollow sites. Also, the adsorbed NO in bridge sites tend to move to the position that is near the surface Sc atom during geometry optimization. The outstanding elongation of NO bond length contributes to change the molecular orbit.
Graphic abstract
中文翻译:
Al基金属间化合物L1 $$ _ {{{2}} $$ 2 -Al $$ _ 3 $$ 3 Sc在(111)表面上没有吸附
摘要
Al基金属间化合物L1 \(_ {\ mathrm {2}} \)- Al \(_ {\ mathrm {3}} \) Sc表现出良好的NO吸附性能,最大吸附能达到近3 eV对应于N–O距离的显着延长(大于20%)。预测结果表明,在0.25的覆盖率下,Al \(_ {\ mathrm {3}} \) Sc(1 1 1)表面的能量有利吸附构型是桥和某些中空部位。而且,在几何优化期间,桥位处吸附的NO趋于移动到靠近表面Sc原子的位置。NO键长度的出色延伸有助于改变分子轨道。








































 京公网安备 11010802027423号
京公网安备 11010802027423号