当前位置:
X-MOL 学术
›
Trans. Emerg. Telecommun. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of protein-protein interactions using stacked auto-encoder
Transactions on Emerging Telecommunications Technologies ( IF 2.5 ) Pub Date : 2021-03-23 , DOI: 10.1002/ett.4256 Kanchan Jha 1 , Sriparna Saha 1 , M. Tanveer 2
Transactions on Emerging Telecommunications Technologies ( IF 2.5 ) Pub Date : 2021-03-23 , DOI: 10.1002/ett.4256 Kanchan Jha 1 , Sriparna Saha 1 , M. Tanveer 2
Affiliation
|
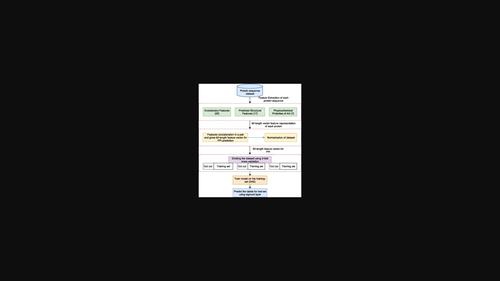
Protein-protein interactions (PPIs) play essential roles in understanding the protein functions and the corresponding pathways which are involved in various biological processes, as well as help in understanding the cause and growth of diseases. Several computational methods such as Support Vector Machine and decision tree are popularly used along with the experimental methods to address the PPIs problem. Such algorithms consider different protein features, including protein sequence, genomes, protein structure, function, topology of the PPIs network, and those that combine multiple aspects. Nowadays, Deep learning (DL) algorithms are successfully used in solving problems in different domains. So, in this paper, we have used stacked auto-encoder as one of the DL methods in solving the problem of PPIs. This model takes the input 92-length feature vector, which is the integration of features extracted from the protein sequence using different methods. The feature vector consists of evolutionary features obtained by PSI-BLAST algorithm, predicted structural properties obtained by SPIDER2, and seven physicochemical properties of amino acids. The key novelty of the current study lies in extracting useful features to solve the PPI problem. The results obtained by our method of feature extraction are compared with those obtained by other feature extraction methods such as Autocovariance and Conjoint-triad, and our proposed feature extraction method is found to be relatively more accurate.
中文翻译:

使用堆叠自动编码器预测蛋白质-蛋白质相互作用
蛋白质-蛋白质相互作用 (PPI) 在了解蛋白质功能和参与各种生物过程的相应途径以及帮助了解疾病的病因和发展方面发挥着重要作用。支持向量机和决策树等几种计算方法与实验方法一起被广泛用于解决 PPI 问题。此类算法考虑了不同的蛋白质特征,包括蛋白质序列、基因组、蛋白质结构、功能、PPIs 网络的拓扑结构,以及结合多个方面的那些。如今,深度学习(DL)算法已成功用于解决不同领域的问题。因此,在本文中,我们使用堆叠自动编码器作为解决 PPI 问题的 DL 方法之一。该模型采用输入的 92 长特征向量,它是使用不同方法从蛋白质序列中提取的特征的整合。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。
更新日期:2021-03-23
中文翻译:
使用堆叠自动编码器预测蛋白质-蛋白质相互作用
蛋白质-蛋白质相互作用 (PPI) 在了解蛋白质功能和参与各种生物过程的相应途径以及帮助了解疾病的病因和发展方面发挥着重要作用。支持向量机和决策树等几种计算方法与实验方法一起被广泛用于解决 PPI 问题。此类算法考虑了不同的蛋白质特征,包括蛋白质序列、基因组、蛋白质结构、功能、PPIs 网络的拓扑结构,以及结合多个方面的那些。如今,深度学习(DL)算法已成功用于解决不同领域的问题。因此,在本文中,我们使用堆叠自动编码器作为解决 PPI 问题的 DL 方法之一。该模型采用输入的 92 长特征向量,它是使用不同方法从蛋白质序列中提取的特征的整合。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。特征向量由PSI-BLAST算法得到的进化特征、SPIDER2得到的预测结构特性和氨基酸的7个理化特性组成。当前研究的关键创新在于提取有用的特征来解决 PPI 问题。将我们的特征提取方法获得的结果与自协方差和联合三元组等其他特征提取方法获得的结果进行比较,发现我们提出的特征提取方法相对更准确。










































 京公网安备 11010802027423号
京公网安备 11010802027423号