The European Physical Journal B ( IF 1.6 ) Pub Date : 2021-03-22 , DOI: 10.1140/epjb/s10051-021-00070-6 T. Guerra , L. R. S. Araújo , S. Azevedo
|
Abstract
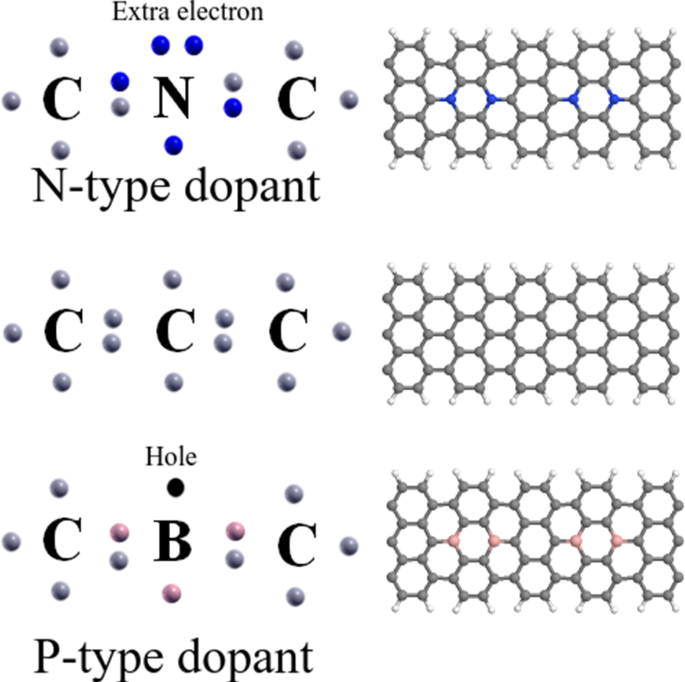
Nanoribbons are strong candidates for use in future nanoelectronics devices due to reduced dimensionality end fascinating properties. Despite their rich properties, they have some limitations such as the wide bandgap and no magnetic order of boron nitride nanoribbons and the absence/small energy gap of graphene nanoribbons. The chemical doping of nanoribbons is an interesting topic since the structural, electronic, magnetic, and quantum transport properties could be significantly altered depending on the species of the dopant, the location of the dopants in the structure and their concentration. Here, we study the chemical doping of armchair and zigzag graphene/boron nitride nanoribbons using density functional theory. We investigate the structural, electronic and magnetic properties of these systems. We find that the armchair edge could reduce its energy by establishing a double bond between the outer carbon atoms. Chemical doping with boron and nitrogen atoms in graphene nanoribbons act as a p-type and n-type dopant, which introduce impurity states close to the valence and conduction bands, increasing/opening the energy gap tuning both the nanoelectronic and the nanomagnetic properties. In boron nitride nanoribbons the controlled chemical doping shows itself an important tool to reduce the wide energy gap and to introduce magnetism in these systems. The zigzag graphene nanoribbons, originally, present an imbalance between spin up and spin down, such as their edges belong to different sublattices. However, the doping, in different sublattices, due to the same type of atom, produces a balance between spin up and spin down, resulting in a null polarization.
Graphic abstract
中文翻译:
石墨烯和氮化硼纳米带的多重掺杂:从头算研究
摘要
纳米带由于减小的尺寸引人入胜的特性而成为用于未来的纳米电子器件的强候选。尽管它们具有丰富的特性,但它们仍具有一些局限性,例如宽带隙宽,氮化硼纳米带没有磁阶,石墨烯纳米带不存在/能隙小。纳米带的化学掺杂是一个有趣的话题,因为结构,电子,磁性和量子传输性质可能会根据掺杂物的种类,掺杂物在结构中的位置及其浓度而发生显着变化。在这里,我们使用密度泛函理论研究扶手椅和锯齿形石墨烯/氮化硼纳米带的化学掺杂。我们研究了这些系统的结构,电子和磁性。我们发现,扶手椅边缘可以通过在外部碳原子之间建立双键来减少其能量。石墨烯纳米带中硼和氮原子的化学掺杂充当p型和n型掺杂剂,它们会引入接近化合价和导带的杂质态,从而增加/打开了能带,从而调节了纳米电子和纳米磁性。在氮化硼纳米带中,受控化学掺杂本身显示出是减小宽能隙并在这些系统中引入磁性的重要工具。之字形石墨烯纳米带最初在向上旋转和向下旋转之间存在不平衡,例如它们的边缘属于不同的亚晶格。但是,由于原子类型相同,在不同的子晶格中进行掺杂会在自旋向上和向下旋转之间产生平衡,










































 京公网安备 11010802027423号
京公网安备 11010802027423号