Journal of Molecular Spectroscopy ( IF 1.4 ) Pub Date : 2021-03-12 , DOI: 10.1016/j.jms.2021.111446 Paul M. Mayer , Andras Bodi
|
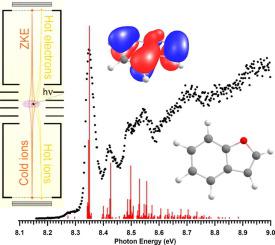
Oxygen-containing polycyclic aromatic hydrocarbons (OPAHs) have been considered as potential carriers of the 11.3 μm band in interstellar observations. To begin to probe their potential contribution to chemistry in the interstellar medium, we report here threshold photoelectron spectra (TPES) for seven oxygen-containing polycyclic aromatic hydrocarbons 2,3-benzofuran (BF), 2,3-dihydrobenzofuran (DHF), phthalan (PH), dibenzofuran (DBF), dibenzo(B,F)oxepine (DBO), benzo[b]naphtho[2,3-d]furan (BNF) and benzo[b]naphtho[1,2-d]furan (cBNF). Vertical ionization energies to the ground and excited ion states were calculated with the outer-valence Green’s function method, and Franck–Condon simulations, based on harmonic (time-dependent) density functional theory results, were used to explore vibrational excitation upon ionization. CBS-QB3 calculations are also reported. Adiabatic ionization energies (IE) could be measured accurately for five of the molecules: BF (8.35 ± 0.01 eV), PH (8.76 ± 0.01 eV), DBF (8.12 ± 0.02 eV), BNF (7.64 ± 0.02 eV), and cBNF (7.62 ± 0.02 eV). For BF and cBNF, excited state optimizations also allowed Franck–Condon simulations of excited-state bands. The TPES of DHF and DBO feature broad ionization onsets due to large geometry changes upon ionization, as both become planar when an electron is removed. The band maxima yield the vertical IE for these two molecules: 8.2 ± 0.2 eV and 8.0 ± 0.2 eV, respectively, while the best estimate of the adiabatic IE for DHF is 7.98 ± 0.05 eV.
中文翻译:
含氧多环芳烃的VUV光处理:阈值光电子能谱
在星际观测中,含氧多环芳烃(OPAH)被认为是11.3μm波段的潜在载体。为了开始探究它们对星际介质中化学的潜在贡献,我们在此报告七种含氧多环芳烃2,3-苯并呋喃(BF),2,3-二氢苯并呋喃(DHF)和邻苯二甲酸的阈值光电子能谱(TPES) (PH),二苯并呋喃(DBF),二苯并(B,F)氧杂平(DBO),苯并[ b ]萘[2,3-d]呋喃(BNF)和苯并[ b]萘[1,2-d]呋喃(cBNF)。利用外价格林函数方法计算了垂直基态的电离能和激发态的电离能,并基于谐波(随时间变化)密度泛函理论的结果,使用Franck-Condon模拟来探索电离时的振动激发。还报告了CBS-QB3的计算。可以精确测量五个分子的绝热电离能(IE):BF(8.35±0.01 eV),PH(8.76±0.01 eV),DBF(8.12±0.02 eV),BNF(7.64±0.02 eV)和cBNF (7.62±0.02 eV)。对于BF和cBNF,激发态优化还允许对激发态谱带进行Franck-Condon模拟。DHF和DBO的TPES具有较大的电离起点,这是由于电离时几何形状发生较大变化,因为当电子被移除时,两者都变为平面。










































 京公网安备 11010802027423号
京公网安备 11010802027423号