当前位置:
X-MOL 学术
›
Soft Matter
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural transitions at electrodes, immersed in simple ionic liquid models
Soft Matter ( IF 2.9 ) Pub Date : 2021-2-11 , DOI: 10.1039/d0sm02167a Hongduo Lu 1, 2, 3, 4, 5 , Samuel Stenberg 1, 2, 3, 4, 5 , Clifford E. Woodward 6, 7, 8, 9, 10 , Jan Forsman 1, 2, 3, 4, 5
Soft Matter ( IF 2.9 ) Pub Date : 2021-2-11 , DOI: 10.1039/d0sm02167a Hongduo Lu 1, 2, 3, 4, 5 , Samuel Stenberg 1, 2, 3, 4, 5 , Clifford E. Woodward 6, 7, 8, 9, 10 , Jan Forsman 1, 2, 3, 4, 5
Affiliation
|
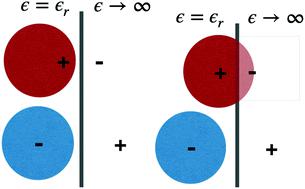
We used a recently developed classical Density Functional Theory (DFT) method to study the structures, phase transitions, and electrochemical behaviours of two coarse-grained ionic fluid models, in the presence of a perfectly conducting model electrode. Common to both is that the charge of the cationic component is able to approach the electrode interface more closely than the anion charge. This means that the cations are specifically attracted to the electrode, due to surface polarization effects. Hence, for a positively charged electrode, there is competition at the surface between cations and anions, where the latter are attracted by the positive electrode charge. This generates demixing, for a range of positive voltages, where the two phases are structurally quite different. The surface charge density is also different between the two phases, even at the same potential. The DFT formulation contains an approximate treatment of ion correlations, and surface polarization, where the latter is modelled via screened image interactions. Using a mean-field DFT, where ion correlations are neglected, causes the phase transition to vanish for both models, but there is still a dramatic drop in the differential capacitance as proximal cations are replaced by anions, for increasing surface potentials. While these findings were obtained for relatively crude coarse-grained models, we argue that the findings can also be relevant in “real” systems, where we note that many ionic liquids are composed of a spherically symmetric anion, and a cation that is asymmetric both from a steric and a charge distribution point of view.
中文翻译:

浸入简单离子液体模型中的电极的结构转变
我们使用一种最新开发的经典密度泛函理论(DFT)方法,在存在完美导电模型电极的情况下,研究了两种粗粒度离子流体模型的结构,相变和电化学行为。两者的共同点是,阳离子组分的电荷比阴离子电荷更接近电极界面。这意味着由于表面极化效应,阳离子被特异性地吸引到电极上。因此,对于带正电的电极,在阳离子和阴离子之间的表面存在竞争,后者被正电荷吸引。对于一定范围的正电压,这会产生混合,其中两相在结构上完全不同。两相之间的表面电荷密度也不同,即使潜力相同。DFT公式包含对离子相关性和表面极化的近似处理,其中对后者进行了建模通过筛选的图像交互。使用忽略离子相关性的均值场DFT会使两个模型的相变消失,但由于近端阳离子被阴离子取代而增加了表面电势,因此差分电容仍然会急剧下降。尽管这些发现是从相对粗略的粗粒度模型获得的,但我们认为这些发现在“真实”系统中也可能是相关的,在该系统中,我们注意到许多离子液体由球形对称的阴离子和不对称的阳离子组成从空间和电荷分布的角度来看。
更新日期:2021-03-04
中文翻译:
浸入简单离子液体模型中的电极的结构转变
我们使用一种最新开发的经典密度泛函理论(DFT)方法,在存在完美导电模型电极的情况下,研究了两种粗粒度离子流体模型的结构,相变和电化学行为。两者的共同点是,阳离子组分的电荷比阴离子电荷更接近电极界面。这意味着由于表面极化效应,阳离子被特异性地吸引到电极上。因此,对于带正电的电极,在阳离子和阴离子之间的表面存在竞争,后者被正电荷吸引。对于一定范围的正电压,这会产生混合,其中两相在结构上完全不同。两相之间的表面电荷密度也不同,即使潜力相同。DFT公式包含对离子相关性和表面极化的近似处理,其中对后者进行了建模通过筛选的图像交互。使用忽略离子相关性的均值场DFT会使两个模型的相变消失,但由于近端阳离子被阴离子取代而增加了表面电势,因此差分电容仍然会急剧下降。尽管这些发现是从相对粗略的粗粒度模型获得的,但我们认为这些发现在“真实”系统中也可能是相关的,在该系统中,我们注意到许多离子液体由球形对称的阴离子和不对称的阳离子组成从空间和电荷分布的角度来看。










































 京公网安备 11010802027423号
京公网安备 11010802027423号