Bioorganic & Medicinal Chemistry ( IF 3.3 ) Pub Date : 2021-02-25 , DOI: 10.1016/j.bmc.2021.116094 Qiao Liu 1 , Yanli Luo 1 , Zerui Li 1 , Chen Chen 2 , Lei Fang 1
|
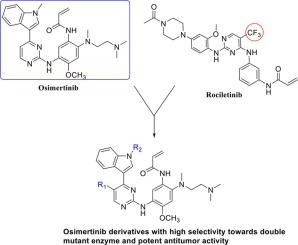
EGFR inhibitors represent a significant milestone for treatment of non-small cell lung cancer, however, they suffer from the acquired drug resistance. Utilizing osimertinib as the lead compound, this work has explored the structural modifications on the indole and pyrimidine rings of osimertinib to generate novel osimertinib derivatives. The in vitro enzymatic and cellular studies showed that the derivatives possessed high selectivity towards double mutant EGFR and potent antitumor activity. Particularly, compound 6b-1, the most active compound, exhibited excellent inhibitory activity against double mutant EGFR (IC50 = 0.18 nM) and wild-type EGFR (IC50 = 2.89 nM) as well as H1975 cells (IC50 = 1.44 nM). Western blot analysis showed that 6b-1 completely inhibited double mutant EGFR and Erk phosphorylation. In vivo test using xenograft model indicated that compound 6b-1 had better antitumor efficacy than osimertinib. More importantly, 6b-1 displayed many advantages in the pharmacokinetic study, including better oral bioavailability and metabolism character.
中文翻译:
osimertinib 吲哚和嘧啶环的结构修饰导致对 L858R/T790M 双突变酶的高选择性和有效的抗肿瘤活性
EGFR抑制剂是治疗非小细胞肺癌的一个重要里程碑,然而,它们存在获得性耐药性。利用奥希替尼作为先导化合物,这项工作探索了奥希替尼的吲哚和嘧啶环的结构修饰,以产生新的奥希替尼衍生物。在体外酶活性和细胞的研究表明,具有衍生物朝双突变的EGFR和有效的抗肿瘤活性的高选择性。特别是,活性最强的化合物6b-1对双突变EGFR(IC 50 = 0.18 nM)和野生型EGFR(IC 50 = 2.89 nM)以及H1975细胞(IC 50 = 1.44 nM)。蛋白质印迹分析表明,6b-1完全抑制双突变 EGFR 和 Erk 磷酸化。使用异种移植模型的体内试验表明,化合物6b-1比奥希替尼具有更好的抗肿瘤功效。更重要的是,6b-1在药代动力学研究中显示出许多优势,包括更好的口服生物利用度和代谢特性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号