当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of dynamic correlation on the ultrafast relaxation of uracil in the gas phase
Faraday Discussions ( IF 3.3 ) Pub Date : 2020-10-30 , DOI: 10.1039/d0fd00110d Pratip Chakraborty 1 , Yusong Liu 2 , Thomas Weinacht 2 , Spiridoula Matsika 1
Faraday Discussions ( IF 3.3 ) Pub Date : 2020-10-30 , DOI: 10.1039/d0fd00110d Pratip Chakraborty 1 , Yusong Liu 2 , Thomas Weinacht 2 , Spiridoula Matsika 1
Affiliation
|
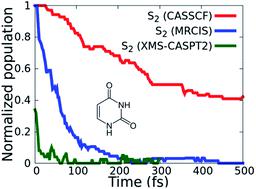
The photophysics and photochemistry of DNA/RNA nucleobases have been extensively investigated during the past two decades, both experimentally and theoretically. The ultrafast relaxation of the canonical nucleobases following photoexcitation is of significant interest when it comes to understanding how nature has ensured their photostability. Here we study the excited state dynamics of uracil which is a nucleobase found in RNA. Although theory and experiment have shed significant light on understanding the photoexcited dynamics of uracil, there are still disagreements in the literature about specific details. In order to examine how the dynamics is influenced by the underlying electronic structure theory, we have performed non-adiabatic excited state dynamics simulations of uracil using on-the-fly trajectory surface hopping methodology on potential energy surfaces calculated at different electronic structure theory levels (CASSCF, MRCIS, XMS-CASPT2, TD-DFT). These simulations reveal that the dynamics are very sensitive to the underlying electronic structure theory, with the multi-reference theory levels that include dynamic correlation, predicting that there is no trapping on the absorbing S2 state, in contrast to predictions from lower level electronic structure results. The dynamics are instead governed by ultrafast decay to the ground state, or trapping on the dark S1 state.
中文翻译:

动态相关性对尿嘧啶在气相中超快弛豫的影响
在过去的二十年里,DNA/RNA 核碱基的光物理学和光化学在实验和理论上都得到了广泛的研究。在了解自然如何确保其光稳定性时,光激发后典型核碱基的超快弛豫具有重要意义。在这里,我们研究了在 RNA 中发现的核碱基尿嘧啶的激发态动力学。尽管理论和实验对理解尿嘧啶的光激发动力学有重要意义,但文献中对具体细节仍存在分歧。为了研究动力学如何受到潜在电子结构理论的影响,我们使用动态轨迹表面跳跃方法对不同电子结构理论水平(CASSCF、MRCIS、XMS-CASPT2、TD-DFT)计算的势能表面进行了尿嘧啶的非绝热激发态动力学模拟。这些模拟表明动力学对潜在的电子结构理论非常敏感,多参考理论水平包括动态相关性,预测吸收的 S 没有陷阱2状态,与来自较低级别电子结构结果的预测相反。相反,动力学由超快衰减到基态或陷入暗 S 1状态来控制。
更新日期:2020-10-30
中文翻译:
动态相关性对尿嘧啶在气相中超快弛豫的影响
在过去的二十年里,DNA/RNA 核碱基的光物理学和光化学在实验和理论上都得到了广泛的研究。在了解自然如何确保其光稳定性时,光激发后典型核碱基的超快弛豫具有重要意义。在这里,我们研究了在 RNA 中发现的核碱基尿嘧啶的激发态动力学。尽管理论和实验对理解尿嘧啶的光激发动力学有重要意义,但文献中对具体细节仍存在分歧。为了研究动力学如何受到潜在电子结构理论的影响,我们使用动态轨迹表面跳跃方法对不同电子结构理论水平(CASSCF、MRCIS、XMS-CASPT2、TD-DFT)计算的势能表面进行了尿嘧啶的非绝热激发态动力学模拟。这些模拟表明动力学对潜在的电子结构理论非常敏感,多参考理论水平包括动态相关性,预测吸收的 S 没有陷阱2状态,与来自较低级别电子结构结果的预测相反。相反,动力学由超快衰减到基态或陷入暗 S 1状态来控制。










































 京公网安备 11010802027423号
京公网安备 11010802027423号