当前位置:
X-MOL 学术
›
React. Chem. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational modelling of adsorption and diffusion properties of CO2 and CH4 in ZIF-8 for gas separation applications: a density functional theory approach
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2021-1-13 , DOI: 10.1039/d0re00416b Hari P. Paudel 1, 2, 3, 4 , Wei Shi 1, 2, 3, 4 , David Hopkinson 1, 2, 3, 4 , Janice A. Steckel 1, 2, 3, 4 , Yuhua Duan 1, 2, 3, 4
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2021-1-13 , DOI: 10.1039/d0re00416b Hari P. Paudel 1, 2, 3, 4 , Wei Shi 1, 2, 3, 4 , David Hopkinson 1, 2, 3, 4 , Janice A. Steckel 1, 2, 3, 4 , Yuhua Duan 1, 2, 3, 4
Affiliation
|
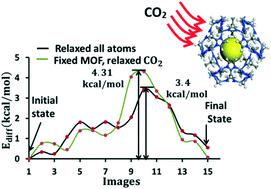
Understanding of zeolitic imidazolate framework-8 (ZIF-8) interaction with different gas molecules is crucial when ZIF-8 is used in gas separation. Computational studies based on density functional theory (DFT) can be used to investigate gas interactions and diffusion mechanisms that can be directly correlated with experimental observations. Here we present our studies based on DFT calculations on CO2 and CH4 gas adsorption and diffusion in the bulk of ZIF-8. We evaluate the structural and electronic properties of bulk ZIF-8, and determine the most stable adsorption sites and the corresponding diffusion barriers for CO2 and CH4 molecules. Our calculations incorporate long-range dispersion interactions to describe the weak interactions between adsorbate molecules and the framework. We analyze the adsorption and diffusion properties in relation with the material's volume expansion. We find that the CO2 and CH4 adsorption energies at the most stable adsorption sites are 5.01 and 4.47 kcal mol−1, respectively. The diffusivity of CO2 is found to be about two times that of CH4. Our calculated diffusion coefficients were found to have the same order of magnitude with the experimental results. Furthermore, our calculations indicate that CO2 and CH4 diffusivities in fixed ZIF-8 (all ZIF-8 framework atoms are fixed) are 5–9 times lower than the corresponding diffusion values in flexible ZIF-8 (all the framework atoms are allowed to move).
中文翻译:

用于气体分离应用的ZIF-8中CO2和CH4的吸附和扩散特性的计算模型:密度泛函理论方法
当在气体分离中使用ZIF-8时,了解沸石咪唑酸酯骨架8(ZIF-8)与不同气体分子的相互作用至关重要。基于密度泛函理论(DFT)的计算研究可用于研究与实验观察结果直接相关的气体相互作用和扩散机理。在这里,我们基于DFT计算对ZIF-8主体中CO 2和CH 4气体的吸附和扩散进行了介绍。我们评估了整体ZIF-8的结构和电子性能,并确定了最稳定的吸附位点和相应的CO 2和CH 4扩散屏障。分子。我们的计算结合了远程分散相互作用,以描述被吸附物分子与骨架之间的弱相互作用。我们分析了与材料体积膨胀有关的吸附和扩散特性。我们发现,最稳定的吸附位处的CO 2和CH 4吸附能分别为5.01和4.47 kcal mol -1。发现CO 2的扩散率约为CH 4的两倍。我们计算出的扩散系数与实验结果具有相同的数量级。此外,我们的计算表明,CO 2和CH 4 固定ZIF-8(所有ZIF-8框架原子都是固定的)中的扩散率比柔性ZIF-8(所有框架原子都允许移动)中相应的扩散值低5-9倍。
更新日期:2021-02-01
中文翻译:
用于气体分离应用的ZIF-8中CO2和CH4的吸附和扩散特性的计算模型:密度泛函理论方法
当在气体分离中使用ZIF-8时,了解沸石咪唑酸酯骨架8(ZIF-8)与不同气体分子的相互作用至关重要。基于密度泛函理论(DFT)的计算研究可用于研究与实验观察结果直接相关的气体相互作用和扩散机理。在这里,我们基于DFT计算对ZIF-8主体中CO 2和CH 4气体的吸附和扩散进行了介绍。我们评估了整体ZIF-8的结构和电子性能,并确定了最稳定的吸附位点和相应的CO 2和CH 4扩散屏障。分子。我们的计算结合了远程分散相互作用,以描述被吸附物分子与骨架之间的弱相互作用。我们分析了与材料体积膨胀有关的吸附和扩散特性。我们发现,最稳定的吸附位处的CO 2和CH 4吸附能分别为5.01和4.47 kcal mol -1。发现CO 2的扩散率约为CH 4的两倍。我们计算出的扩散系数与实验结果具有相同的数量级。此外,我们的计算表明,CO 2和CH 4 固定ZIF-8(所有ZIF-8框架原子都是固定的)中的扩散率比柔性ZIF-8(所有框架原子都允许移动)中相应的扩散值低5-9倍。










































 京公网安备 11010802027423号
京公网安备 11010802027423号