Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2021-01-29 , DOI: 10.1016/j.jmgm.2021.107848 Andrew E Sifain 1 , Betsy M Rice 1 , Samuel H Yalkowsky 2 , Brian C Barnes 1
|
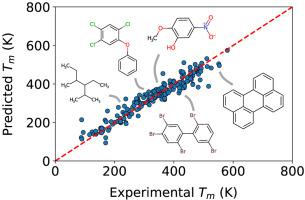
A priori knowledge of physicochemical properties such as melting and boiling could expedite materials discovery. However, theoretical modeling from first principles poses a challenge for efficient virtual screening of potential candidates. As an alternative, the tools of data science are becoming increasingly important for exploring chemical datasets and predicting material properties. Herein, we extend a molecular representation, or set of descriptors, first developed for quantitative structure-property relationship modeling by Yalkowsky and coworkers known as the Unified Physicochemical Property Estimation Relationships (UPPER). This molecular representation has group-constitutive and geometrical descriptors that map to enthalpy and entropy; two thermodynamic quantities that drive thermal phase transitions. We extend the UPPER representation to include additional information about sp2-bonded fragments. Additionally, instead of using the UPPER descriptors in a series of thermodynamically-inspired calculations, as per Yalkowsky, we use the descriptors to construct a vector representation for use with machine learning techniques. The concise and easy-to-compute representation, combined with a gradient-boosting decision tree model, provides an appealing framework for predicting experimental transition temperatures in a diverse chemical space. An application to energetic materials shows that the method is predictive, despite a relatively modest energetics reference dataset. We also report competitive results on diverse public datasets of melting points (i.e., OCHEM, Enamine, Bradley, and Bergström) comprised of over 47k structures. Open source software is available at https://github.com/USArmyResearchLab/ARL-UPPER.
中文翻译:
二维结构的机器学习转变温度
先验物理化学性质(例如熔化和煮沸)的知识可以加快材料发现的速度。然而,基于第一原理的理论建模对有效虚拟筛选潜在候选人提出了挑战。作为替代方案,数据科学的工具对于探索化学数据集和预测材料特性变得越来越重要。在这里,我们扩展了分子表示法或一组描述符,这是由Yalkowsky及其同事首先为定量结构-性质关系建模而开发的,被称为统一物理化学性质估计关系(UPPER)。这种分子表示具有映射到焓和熵的基团本构和几何描述子。驱动热相变的两个热力学量。sp 2-键合的片段。此外,按照Yalkowsky的定义,不是在一系列热力学启发的计算中使用UPPER描述符,而是使用描述符构造用于机器学习技术的矢量表示。简洁,易于计算的表示法与增强梯度的决策树模型相结合,为在各种化学空间中预测实验转变温度提供了一个引人入胜的框架。含能材料的应用表明,尽管含能参考数据集相对较少,但该方法是可预测的。我们还在各种公开的熔点数据集(即,OCHEM,Enamine,Bradley和Bergström)由47k多个结构组成。开源软件可从https://github.com/USArmyResearchLab/ARL-UPPER获得。










































 京公网安备 11010802027423号
京公网安备 11010802027423号