当前位置:
X-MOL 学术
›
J. Phys. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: A molecular electron density theory perspective
Journal of Physical Organic Chemistry ( IF 1.9 ) Pub Date : 2021-01-28 , DOI: 10.1002/poc.4189 Nivedita Acharjee 1 , Haydar A. Mohammad‐Salim 2 , Mrinmoy Chakraborty 3 , Madhuri P. Rao 4 , Madhu Ganesh 4, 5
Journal of Physical Organic Chemistry ( IF 1.9 ) Pub Date : 2021-01-28 , DOI: 10.1002/poc.4189 Nivedita Acharjee 1 , Haydar A. Mohammad‐Salim 2 , Mrinmoy Chakraborty 3 , Madhuri P. Rao 4 , Madhu Ganesh 4, 5
Affiliation
|
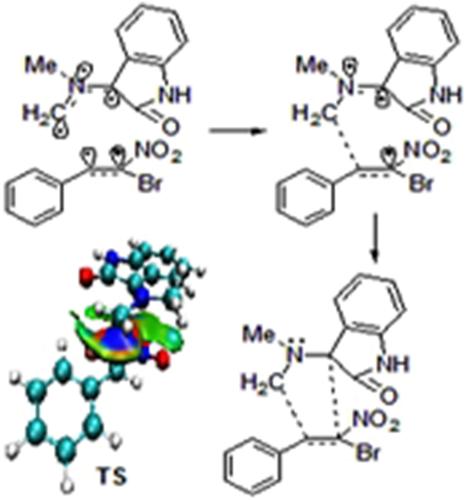
A molecular electron density theory (MEDT) study for the [3 + 2] cycloaddition (32CA) reaction of indoledione and N‐methyl glycine with (E)‐1‐bromonitrostyrene leading to the spirooxindole‐pyrrolidine adduct is presented. Electron localisation function (ELF) study of the in situ generated azomethine ylide classifies a pseudodiradical electronic structure associated with low activation energies. This 32CA reaction is a polar process with electronic flux from the strongly nucleophilic azomethine ylide to the strongly electrophilic nitrostyrene, confirmed from the global electron density transfer (GEDT) above 0.20 e at the transition states (TSs). Parr function analysis predicts the correct regioselectivity from the two‐centre interactions. The reaction is kinetically controlled with negative free energy of reaction which makes it irreversible. The activation enthalpy of the favoured TS is lowered by 3.4, 6.3 and 9.3 kcal mol−1 in methanol relative to the other feasible reaction paths, consistent with the experimentally observed complete regioselectivity and stereoselectivity. ELF topological characterisation along the reaction path predicts a two‐stage one‐step molecular mechanism, and bond formation is controlled by the nucleophilic character of the azomethine ylide. ELF study, Laplacian of electron density and independent gradient model (IGM) analysis predict non‐covalent interactions at the TSs, where the new C–C bond formation has not been started.
中文翻译:

螺氧并吲哚并吡咯烷合成中具有高区域选择性和立体选择性的分子电子密度理论研究
提出了一种分子电子密度理论(MEDT),用于吲哚二酮和N-甲基甘氨酸与(E)-1-溴硝基苯乙烯的[3 + 2]环加成(32CA)反应,导致螺环吲哚-吡咯烷加合物的生成。原位产生的甲亚胺叶立德的电子定位功能(ELF)研究将伪双自由基分类与低活化能相关的电子结构。此32CA反应是一个极性过程,其中电子从强亲核甲亚胺叶立德流向强亲电子硝基苯乙烯,这在过渡态(TSs)处的全局电子密度转移(GEDT)大于0.20 e时得到了证实。Parr功能分析可通过两中心相互作用预测正确的区域选择性。反应是由负的反应自由能动力学控制的,这使其不可逆。有利的TS的活化焓降低了3.4、6.3和9.3kcal mol -1在甲醇中相对于其他可行的反应路径,与实验观察到的完全区域选择性和立体选择性一致。沿着反应路径的ELF拓扑表征预测了一个两阶段的一步式分子机理,并且键的形成受偶氮甲内酯的亲核特性控制。ELF研究,电子密度的拉普拉斯算子和独立梯度模型(IGM)分析预测TS处的非共价相互作用,而尚未开始新的C–C键形成。
更新日期:2021-01-28
中文翻译:
螺氧并吲哚并吡咯烷合成中具有高区域选择性和立体选择性的分子电子密度理论研究
提出了一种分子电子密度理论(MEDT),用于吲哚二酮和N-甲基甘氨酸与(E)-1-溴硝基苯乙烯的[3 + 2]环加成(32CA)反应,导致螺环吲哚-吡咯烷加合物的生成。原位产生的甲亚胺叶立德的电子定位功能(ELF)研究将伪双自由基分类与低活化能相关的电子结构。此32CA反应是一个极性过程,其中电子从强亲核甲亚胺叶立德流向强亲电子硝基苯乙烯,这在过渡态(TSs)处的全局电子密度转移(GEDT)大于0.20 e时得到了证实。Parr功能分析可通过两中心相互作用预测正确的区域选择性。反应是由负的反应自由能动力学控制的,这使其不可逆。有利的TS的活化焓降低了3.4、6.3和9.3kcal mol -1在甲醇中相对于其他可行的反应路径,与实验观察到的完全区域选择性和立体选择性一致。沿着反应路径的ELF拓扑表征预测了一个两阶段的一步式分子机理,并且键的形成受偶氮甲内酯的亲核特性控制。ELF研究,电子密度的拉普拉斯算子和独立梯度模型(IGM)分析预测TS处的非共价相互作用,而尚未开始新的C–C键形成。










































 京公网安备 11010802027423号
京公网安备 11010802027423号