Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy ( IF 4.3 ) Pub Date : 2021-01-29 , DOI: 10.1016/j.saa.2021.119503 Feixiang Ji , Yurong Guo , Mengqi Wang , Zibo Wu , Yanan Shi , Xiaoying Zhao , Haiyuan Wang , Xia Feng , Guangjiu Zhao
|
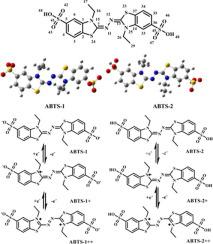
The molecular structures of 2,2-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), were calculated by using time-dependent density functional theory (TDDFT) model with M062X method with 6-311G (d, p) basis set. In this work, the ABTS were theoretically investigated from the geometric structure, the energy levels of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO), the energy level gap ΔEHOMO-LUMO of the molecular ground state, excited stated properties and the electronic absorption spectra of different oxidation states. We studied the energy levels of LUMO and HOMO of ABTS in different oxidation states. Frontier molecular orbital analysis can provide insight into the nature of excited states. ABTS was synthesized from N-ethylamine by total synthesis. Then, we measured the UV–Vis spectra of ABTS before and after being oxidized by K2S2O8. The calculated electronic structures and photochemical properties of different oxidation state of ABTS were in accordance with the experimental result. This work demonstrates the relationship between the electronic structures and photochemistry of different oxidation states ABTS hence paves the way for the rationally synthesis and deepen understanding of the photophysical properties of ABTS materials.
中文翻译:
2,2'-叠氮基双-(3-乙基苯并噻唑啉-6-磺酸)(ABTS)不同氧化态的激发态电子结构和光化学
使用M062X方法和时间依赖性密度泛函理论(TDDFT)模型,以6-311G(d,2,2-叠氮基-双-(3-乙基苯并噻唑啉-6-磺酸)(ABTS)的分子结构进行计算。 p)基础集。在这项工作中,从几何结构,最低未占据分子轨道(LUMO)和最高占据分子轨道(HOMO)的能级,分子基态的能级间隙ΔE HOMO-LUMO的角度对ABTS进行了理论研究,激发态性质和不同氧化态的电子吸收光谱。我们研究了不同氧化态下ABTS的LUMO和HOMO的能级。前沿分子轨道分析可以洞悉激发态的性质。ABTS由N合成-乙胺全合成。然后,我们测量了ABTS被K 2 S 2 O 8氧化前后的UV-Vis光谱。计算得到的ABTS不同氧化态的电子结构和光化学性质与实验结果吻合。这项工作证明了不同氧化态ABTS的电子结构与光化学之间的关系,从而为合理合成和加深对ABTS材料的光物理性质的铺平了道路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号