当前位置:
X-MOL 学术
›
Phys. Rev. Materials
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Anomalous electronic properties in layered, disordered ZnVSb
Physical Review Materials ( IF 3.1 ) Pub Date : 2021-01-27 , DOI: 10.1103/physrevmaterials.5.015002 Erik A. Bensen , Kamil Ciesielski , Lídia C. Gomes , Brenden R. Ortiz , Johannes Falke , Orest Pavlosiuk , Daniel Weber , Tara L. Braden , Kenneth X. Steirer , Damian Szymański , Joshua E. Goldberger , Chang-Yang Kuo , Chien-Te Chen , Chun-Fu Chang , Liu Hao Tjeng , Dariusz Kaczorowski , Elif Ertekin , Eric S. Toberer
Physical Review Materials ( IF 3.1 ) Pub Date : 2021-01-27 , DOI: 10.1103/physrevmaterials.5.015002 Erik A. Bensen , Kamil Ciesielski , Lídia C. Gomes , Brenden R. Ortiz , Johannes Falke , Orest Pavlosiuk , Daniel Weber , Tara L. Braden , Kenneth X. Steirer , Damian Szymański , Joshua E. Goldberger , Chang-Yang Kuo , Chien-Te Chen , Chun-Fu Chang , Liu Hao Tjeng , Dariusz Kaczorowski , Elif Ertekin , Eric S. Toberer
|
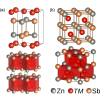
New materials discovery is the driving force for progress in solid state physics and chemistry. Here we solve the crystal structure and comprehensively study physical properties of ZnVSb in the polycrystalline form. Synchrotron x-ray diffraction reveals that the compound attains a layered ZrSiS-type structure (/nmm, = 4.09021(2) Å, = 6.42027(4) Å). The unit cell is composed of a 2D vanadium network separated by Zn-Sb blocks that are slightly distorted from the ideal cubic arrangement. A considerable amount of vacancies were observed on the vanadium and antimony positions; the experimental composition is . Low-temperature x-ray diffraction shows very subtle discontinuity in the lattice parameters around 175 K. Bonding V-V distance is below the critical separation of 2.97 Å known from the literature, which allows for V-V orbital overlap and subsequent metallic conductivity. From ab initio calculations, we found that ZnVSb is a pseudogap material with an expected dominant vanadium contribution to the density of states at the Fermi level. The energy difference between the antiferromagnetic and nonordered magnetic configurations is rather small (0.34 eV/f.u.). X-ray photoelectron spectroscopy fully corroborates the results of the band structure calculations. Magnetic susceptibility uncovered that, in ZnVSb, itinerant charge carriers coexist with a small, localized magnetic moment of ca. 0.25 . The itinerant electrons arise from the ordered part of the vanadium lattice. Fractional localization, in turn, was attributed to V atoms neighboring vacancies, which hinder full V-V orbital overlap. Furthermore, the susceptibility and electrical resistivity showed a large hysteresis between 120 K and 160 K. The effect is not sensitive to magnetic fields up to 9 T. Curie-Weiss fitting revealed that the amount of itinerant charge carriers in ZnVSb drops with decreasing temperature below 160 K, which is accompanied by a concurrent rise in the localized magnetic moment. This observation together with the overall shape of the hysteresis in the resistivity allows for suggesting a plausible origin of the effect as a charge-transfer metal-insulator transition. Ab initio phonon calculations show the formation of a collective phonon mode at 2.8 THz (134 K). The experimental heat capacity reflected this feature by a boson peak with Einstein temperature of 116 K. Analysis of the heat capacity with both an ab initio perspective and Debye-Einstein model revealed a sizable anharmonic contribution to heat capacity, in line with disordered nature of the material. Further investigation of the electron and phonon properties for ZnVSb is likely to provide valuable insight into the relation between structural disorder and the physical properties of transition-metal-bearing compounds.
中文翻译:

层状无序ZnVSb中的异常电子性质
新材料的发现是固态物理学和化学进步的动力。在这里,我们解决晶体结构并全面研究多晶形式的ZnVSb的物理性质。同步加速器X射线衍射表明该化合物具有层状ZrSiS型结构(/ nmm, = 4.09021(2)Å, = 6.42027(4)Å)。晶胞由2D钒网络组成,该网络由Zn-Sb块分隔开,而Zn-Sb块与理想的立方排列略有不同。在钒和锑位置上观察到大量的空位;实验组成是。低温X射线衍射在175 K附近显示出非常微妙的晶格参数不连续性。键合VV距离低于文献中已知的2.97Å的临界间隔,这允许VV轨道重叠和随后的金属导电性。通过从头算计算,我们发现ZnVSb是一种伪能隙材料,钒对费米能级的态密度有预期的贡献。反铁磁结构和无序磁结构之间的能量差很小(0.34 eV / fu)。X射线光电子能谱完全证实了能带结构的计算结果。磁化率表明,在ZnVSb中,流动电荷载流子与大约一个小的局部磁矩共存。0.25。巡回电子来自钒晶格的有序部分。反过来,部分定位归因于V原子相邻的空位,这阻碍了VV轨道的完全重叠。此外,磁化率和电阻率在120 K和160 K之间显示出较大的磁滞。该效应对高达9 T的磁场不敏感。居里-威斯拟合显示,ZnVSb中的流动电荷载流子的数量随温度降低而降低160 K,伴随着局部磁矩的同时上升。该观察结果以及电阻率中的磁滞现象的整体形状允许暗示该效应的合理来源是电荷转移金属-绝缘体的转变。从头算声子计算显示了在2.8 THz(134 K)时形成的集体声子模式。实验热容量通过爱因斯坦温度为116 K的玻色子峰反映了这一特征。从头算角度和Debye-Einstein模型对热容量的分析均显示出对热容量的相当大的非谐贡献,这与热电偶的无序性质相符。材料。ZnVSb的电子和声子特性的进一步研究可能会提供有价值的见解,以了解结构无序与含过渡金属的化合物的物理特性之间的关系。
更新日期:2021-01-27
中文翻译:
层状无序ZnVSb中的异常电子性质
新材料的发现是固态物理学和化学进步的动力。在这里,我们解决晶体结构并全面研究多晶形式的ZnVSb的物理性质。同步加速器X射线衍射表明该化合物具有层状ZrSiS型结构(/ nmm, = 4.09021(2)Å, = 6.42027(4)Å)。晶胞由2D钒网络组成,该网络由Zn-Sb块分隔开,而Zn-Sb块与理想的立方排列略有不同。在钒和锑位置上观察到大量的空位;实验组成是。低温X射线衍射在175 K附近显示出非常微妙的晶格参数不连续性。键合VV距离低于文献中已知的2.97Å的临界间隔,这允许VV轨道重叠和随后的金属导电性。通过从头算计算,我们发现ZnVSb是一种伪能隙材料,钒对费米能级的态密度有预期的贡献。反铁磁结构和无序磁结构之间的能量差很小(0.34 eV / fu)。X射线光电子能谱完全证实了能带结构的计算结果。磁化率表明,在ZnVSb中,流动电荷载流子与大约一个小的局部磁矩共存。0.25。巡回电子来自钒晶格的有序部分。反过来,部分定位归因于V原子相邻的空位,这阻碍了VV轨道的完全重叠。此外,磁化率和电阻率在120 K和160 K之间显示出较大的磁滞。该效应对高达9 T的磁场不敏感。居里-威斯拟合显示,ZnVSb中的流动电荷载流子的数量随温度降低而降低160 K,伴随着局部磁矩的同时上升。该观察结果以及电阻率中的磁滞现象的整体形状允许暗示该效应的合理来源是电荷转移金属-绝缘体的转变。从头算声子计算显示了在2.8 THz(134 K)时形成的集体声子模式。实验热容量通过爱因斯坦温度为116 K的玻色子峰反映了这一特征。从头算角度和Debye-Einstein模型对热容量的分析均显示出对热容量的相当大的非谐贡献,这与热电偶的无序性质相符。材料。ZnVSb的电子和声子特性的进一步研究可能会提供有价值的见解,以了解结构无序与含过渡金属的化合物的物理特性之间的关系。










































 京公网安备 11010802027423号
京公网安备 11010802027423号