当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The hierarchy of ab initio and DFT methods for describing an intramolecular non-covalent Si⋯N contact in the silicon compounds using electron diffraction geometries
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-1-4 , DOI: 10.1039/d0cp05872f Elena F. Belogolova 1, 2, 3, 4 , Sergey A. Shlykov 4, 5, 6 , Alexey V. Eroshin 4, 5, 6 , Evgeniya P. Doronina 1, 2, 3, 4 , Valery F. Sidorkin 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-1-4 , DOI: 10.1039/d0cp05872f Elena F. Belogolova 1, 2, 3, 4 , Sergey A. Shlykov 4, 5, 6 , Alexey V. Eroshin 4, 5, 6 , Evgeniya P. Doronina 1, 2, 3, 4 , Valery F. Sidorkin 1, 2, 3, 4
Affiliation
|
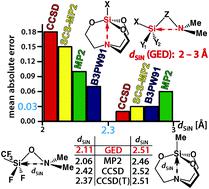
In the series of silatranes XSi(OCH2CH2)3N, 1 (X = Me, 1a; H, 1b; F, 1c) with the known gas electron diffraction (GED) structures, the problematic geometry of 1-methylsilatrane 1a has been revised. In particular, the new value of the Si⋯N distance (dSiN) in 1a turned out to be ∼0.06 Å longer than the generally accepted one. This dSiN resolves the long-standing contradiction between the data of the structural and spectral experiments regarding the sensitivity of 1 to the medium effect. We also performed the ab initio and DFT study of the combined series of silatranes 1a–c, silylalkylamines H3Si(CH2)3NMe2 (2a) and F3SiCH2NMe2 (2b), silylhydrazines F3SiN(Me)NMe2 (2c) and F3SiN(SiMe3)NMe2 (2d), and silyloxyamines ClH2SiONMe2 (2e,f), (F3C)F2SiONMe2 (2g,h) and F3SiONMe2 (2i), in which the GED dSiN values are in a wide range of 2–3 Å. None of the involved quantum chemical methods has succeeded in reproducing all the experimental gas-phase dSiN values in 1a–c, 2a–i with an acceptable accuracy (0.01–0.03 Å). The problems of the used methods, primarily CCSD with the Pople basis sets, are caused by four molecules with the geminal SiNN and SiON fragments (2d,f–i) and dSiN < 2.3 Å. A reasonable hierarchy of computationally accessible theory levels for studying the physicochemical manifestation of the non-covalent intramolecular Si⋯N interactions can be constructed only at dSiN > 2.3 Å: MP2 < PBE0 ∼ B3PW91 ∼ SCS-MP2 < CCSD < CCSD(T).
中文翻译:

从头算和DFT方法的层次结构,用于使用电子衍射几何学描述硅化合物中的分子内非共价Si⋯N接触
在具有已知气体电子衍射(GED)结构的一系列Silatranes XSi(OCH 2 CH 2)3 N,1(X = Me,1a ; H,1b ; F,1c)中,存在1-甲基硅环烷1a的几何问题已修改。特别是,1a中Si⋯N距离(d SiN)的新值比一般公认的值长约0.06Å。此d SiN解决了结构和光谱实验数据之间关于1灵敏度的长期矛盾。达到中等效果。我们还进行了从头组合串联silatranes的和DFT研究1A-1C,silylalkylaminesħ 3的Si(CH 2)3 NME 2(图2a)和F 3 SICH 2 NME 2(图2b),silylhydrazines˚F 3的SiN(ME )NMe 2(2c)和F 3 SiN(SiMe 3)NMe 2(2d)以及甲硅烷氧基胺ClH 2 SiONMe 2(2e,f),(F 3C)F 2 SiONMe 2(2g,h)和F 3 SiONMe 2(2i),其中GED d SiN值在2-3Å的宽范围内。所涉及的量子化学方法均未成功以可接受的精度(0.01-0.03Å)再现1a–c,2a–i中的所有实验气相d SiN值。使用的方法的问题(主要是具有Pople基集的CCSD)是由四个具有双子SiNN和SiON片段(2d,f–i)和d SiN的分子引起的<2.3Å。仅当d SiN > 2.3Å时,才能构建用于研究非共价分子内Si⋯N相互作用的物理化学表现的合理计算层次的理论层次:MP2 <PBE0〜B3PW91〜SCS-MP2 <CCSD <CCSD(T) 。
更新日期:2021-01-26
中文翻译:
从头算和DFT方法的层次结构,用于使用电子衍射几何学描述硅化合物中的分子内非共价Si⋯N接触
在具有已知气体电子衍射(GED)结构的一系列Silatranes XSi(OCH 2 CH 2)3 N,1(X = Me,1a ; H,1b ; F,1c)中,存在1-甲基硅环烷1a的几何问题已修改。特别是,1a中Si⋯N距离(d SiN)的新值比一般公认的值长约0.06Å。此d SiN解决了结构和光谱实验数据之间关于1灵敏度的长期矛盾。达到中等效果。我们还进行了从头组合串联silatranes的和DFT研究1A-1C,silylalkylaminesħ 3的Si(CH 2)3 NME 2(图2a)和F 3 SICH 2 NME 2(图2b),silylhydrazines˚F 3的SiN(ME )NMe 2(2c)和F 3 SiN(SiMe 3)NMe 2(2d)以及甲硅烷氧基胺ClH 2 SiONMe 2(2e,f),(F 3C)F 2 SiONMe 2(2g,h)和F 3 SiONMe 2(2i),其中GED d SiN值在2-3Å的宽范围内。所涉及的量子化学方法均未成功以可接受的精度(0.01-0.03Å)再现1a–c,2a–i中的所有实验气相d SiN值。使用的方法的问题(主要是具有Pople基集的CCSD)是由四个具有双子SiNN和SiON片段(2d,f–i)和d SiN的分子引起的<2.3Å。仅当d SiN > 2.3Å时,才能构建用于研究非共价分子内Si⋯N相互作用的物理化学表现的合理计算层次的理论层次:MP2 <PBE0〜B3PW91〜SCS-MP2 <CCSD <CCSD(T) 。










































 京公网安备 11010802027423号
京公网安备 11010802027423号