当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Extending the Depth of Human Plasma Proteome Coverage Using Simple Fractionation Techniques
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2021-01-20 , DOI: 10.1021/acs.jproteome.0c00670 Gurjeet Kaur 1, 2 , Anne Poljak 1, 2 , Syed Azmal Ali 3 , Ling Zhong 2 , Mark J Raftery 2 , Perminder Sachdev 1, 4
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2021-01-20 , DOI: 10.1021/acs.jproteome.0c00670 Gurjeet Kaur 1, 2 , Anne Poljak 1, 2 , Syed Azmal Ali 3 , Ling Zhong 2 , Mark J Raftery 2 , Perminder Sachdev 1, 4
Affiliation
|
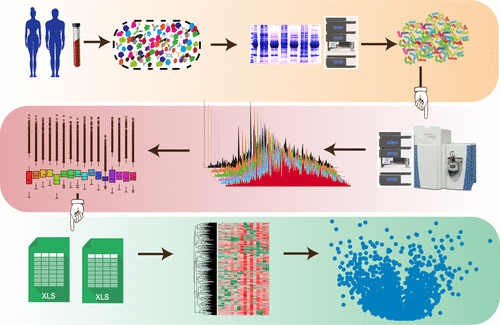
Human plasma is one of the most widely used tissues in clinical analysis, and plasma-based biomarkers are used for monitoring patient health status and/or response to medical treatment to avoid unnecessary invasive biopsy. Data-driven plasma proteomics has suffered from a lack of throughput and detection sensitivity, largely due to the complexity of the plasma proteome and in particular the enormous quantitative dynamic range, estimated to be between 9 and 13 orders of magnitude between the lowest and the highest abundance protein. A major challenge is to identify workflows that can achieve depth of plasma proteome coverage while minimizing the complexity of the sample workup and maximizing the sample throughput. In this study, we have performed intensive depletion of high-abundant plasma proteins or enrichment of low-abundant proteins using the Agilent multiple affinity removal liquid chromatography (LC) column—Human 6 (Hu6), the Agilent multiple affinity removal LC column—Human 14 (Hu14), and ProteoMiner followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE) and C18 prefractionation techniques. We compared the performance of each of these fractionation approaches to identify the method that satisfies requirements for analysis of clinical samples and to include good plasma proteome coverage in combination with reasonable sample output. In this study, we report that one-dimensional (1D) gel-based prefractionation allows parallel sample processing and no loss of proteome coverage, compared with serial chromatographic separation, and significantly accelerates analysis time, particularly important for large clinical projects. Furthermore, we show that a variety of methodologies can achieve similarly high plasma proteome coverage, allowing flexibility in method selection based on project-specific needs. These considerations are important in the effort to accelerate plasma proteomics research so as to provide efficient, reliable, and accurate diagnoses, population-based health screening, clinical research studies, and other clinical work.
中文翻译:

使用简单的分级分离技术扩展人类血浆蛋白质组覆盖的深度
人血浆是临床分析中使用最广泛的组织之一,基于血浆的生物标记物用于监测患者的健康状况和/或对医学治疗的反应,以避免不必要的侵入性活检。数据驱动的血浆蛋白质组学缺乏通量和检测灵敏度,这主要是由于血浆蛋白质组学的复杂性,尤其是巨大的定量动态范围,估计其最低和最高之间在9到13个数量级之间。丰富的蛋白质。一个主要的挑战是确定能够实现血浆蛋白质组覆盖深度的工作流程,同时最大程度地减少样品处理的复杂性并最大程度地提高样品通量。在这个研究中,我们使用安捷伦多亲和力去除液相色谱(LC)色谱柱-Human 6(Hu6),安捷伦多亲和力去除LC色谱柱-Human 14(Hu14)对高丰度血浆蛋白进行了密集消耗或对低丰度蛋白进行了富集和ProteoMiner,然后进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS PAGE)和C18预分离技术。我们比较了每种分馏方法的性能,以确定可满足临床样品分析要求的方法,并包括良好的血浆蛋白质组覆盖率和合理的样品输出量。在这项研究中,我们报告说,与串行色谱分离相比,基于一维(1D)凝胶的预分离技术允许并行处理样品且不会损失蛋白质组覆盖率,并大大缩短了分析时间,这对于大型临床项目尤其重要。此外,我们证明了各种方法都可以实现类似的高血浆蛋白质组覆盖率,从而可以根据项目的特定需求灵活地选择方法。这些考虑因素对加速血浆蛋白质组学研究以提供有效,可靠和准确的诊断,基于人群的健康筛查,临床研究以及其他临床工作至关重要。
更新日期:2021-02-05
中文翻译:
使用简单的分级分离技术扩展人类血浆蛋白质组覆盖的深度
人血浆是临床分析中使用最广泛的组织之一,基于血浆的生物标记物用于监测患者的健康状况和/或对医学治疗的反应,以避免不必要的侵入性活检。数据驱动的血浆蛋白质组学缺乏通量和检测灵敏度,这主要是由于血浆蛋白质组学的复杂性,尤其是巨大的定量动态范围,估计其最低和最高之间在9到13个数量级之间。丰富的蛋白质。一个主要的挑战是确定能够实现血浆蛋白质组覆盖深度的工作流程,同时最大程度地减少样品处理的复杂性并最大程度地提高样品通量。在这个研究中,我们使用安捷伦多亲和力去除液相色谱(LC)色谱柱-Human 6(Hu6),安捷伦多亲和力去除LC色谱柱-Human 14(Hu14)对高丰度血浆蛋白进行了密集消耗或对低丰度蛋白进行了富集和ProteoMiner,然后进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS PAGE)和C18预分离技术。我们比较了每种分馏方法的性能,以确定可满足临床样品分析要求的方法,并包括良好的血浆蛋白质组覆盖率和合理的样品输出量。在这项研究中,我们报告说,与串行色谱分离相比,基于一维(1D)凝胶的预分离技术允许并行处理样品且不会损失蛋白质组覆盖率,并大大缩短了分析时间,这对于大型临床项目尤其重要。此外,我们证明了各种方法都可以实现类似的高血浆蛋白质组覆盖率,从而可以根据项目的特定需求灵活地选择方法。这些考虑因素对加速血浆蛋白质组学研究以提供有效,可靠和准确的诊断,基于人群的健康筛查,临床研究以及其他临床工作至关重要。










































 京公网安备 11010802027423号
京公网安备 11010802027423号