Interdisciplinary Sciences: Computational Life Sciences ( IF 3.9 ) Pub Date : 2021-01-21 , DOI: 10.1007/s12539-020-00411-6 Hai-Yun Wang 1 , Jian-Ping Zhao 1, 2 , Chun-Hou Zheng 1, 3
|
Abstract
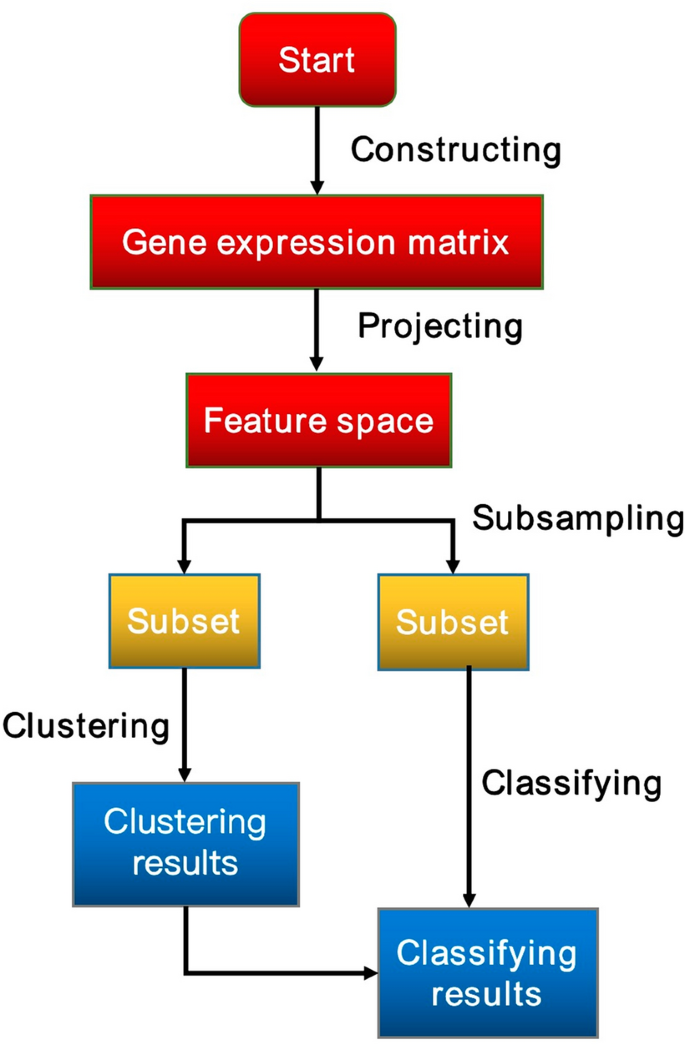
Clustering is a common method to identify cell types in single cell analysis, but the increasing size of scRNA-seq datasets brings challenges to single cell clustering. Therefore, it is an urgent need to design a faster and more accurate clustering method for large-scale scRNA-seq data. In this paper, we proposed a new method for single cell clustering. First, a count matrix is constructed through normalization and gene filtration. Second, the raw data of gene expression matrix are projected to feature space constructed by secondary construction of feature space based on UMAP (Uniform Manifold Approximation and Projection). Third, the low-dimensional matrix on the feature space is randomly divided into two sub-matrices according to a certain proportion for clustering and classifying, respectively. Finally, one subset is clustered by k-means algorithm and then the other subset is classified by k-nearest neighbor algorithm based on clustering results. Experimental results show that our method can cluster the scRNA-seq datasets effectively.
Graphical abstract
中文翻译:
SUSCC:基于UMAP的特征空间二次构建,用于快速准确聚类大规模单细胞RNA-seq数据
摘要
聚类是单细胞分析中识别细胞类型的常用方法,但越来越大的scRNA-seq数据集给单细胞聚类带来了挑战。因此,迫切需要为大规模scRNA-seq数据设计一种更快、更准确的聚类方法。在本文中,我们提出了一种新的单细胞聚类方法。首先,通过归一化和基因过滤构建计数矩阵。其次,将基因表达矩阵的原始数据投影到基于UMAP(Uniform Manifold Approximation and Projection)二次构建特征空间构建的特征空间。第三,将特征空间上的低维矩阵按一定比例随机分成两个子矩阵,分别进行聚类和分类。最后,一个子集通过k-means算法进行聚类,然后另一个子集根据聚类结果通过k-最近邻算法进行分类。实验结果表明,我们的方法可以有效地对 scRNA-seq 数据集进行聚类。










































 京公网安备 11010802027423号
京公网安备 11010802027423号