The Journal of Membrane Biology ( IF 2.3 ) Pub Date : 2021-01-20 , DOI: 10.1007/s00232-020-00167-6 Khuraijam Dhanachandra Singh 1 , Sadashiva S Karnik 1
|
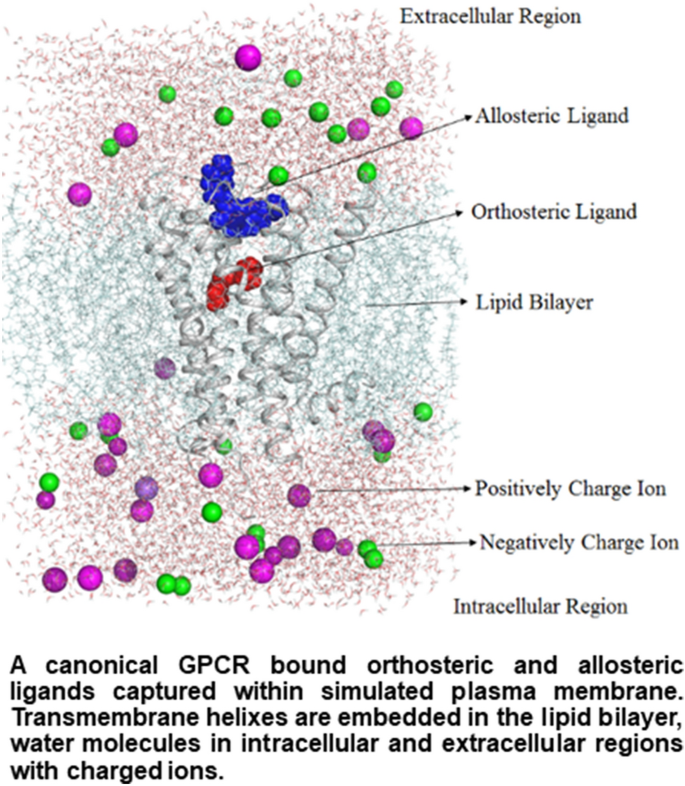
GPCRs remain the most important drug target comprising ~ 34% of the Food and Drug Administration (FDA)-approved drugs. In modern pharmacology of GPCRs, modulating receptor signaling based on requirement of a specific disorder is of immense interest. Classical drugs targeting orthosteric sites in GPCRs completely block the binding of endogenous ligand and consequently inhibit all important signals from a GPCR. Some of many signals elicited by the endogenous ligands may play vital role and inhibiting these may also cause severe side effects in the long run. However, allosteric drugs can modulate GPCR signaling without blocking the endogenous ligand binding. Therefore, allosteric drugs can maintain beneficial signaling of the receptor and prevent unwanted side effects. In this chapter, we will discuss GPCR crystal structures solved with allosteric ligands, advantages of allosteric drugs, and allosteric drugs which are in clinical use or trials.
Graphic Abstract
中文翻译:
GPCR 变构的当前趋势
GPCR 仍然是最重要的药物靶点,约占美国食品药品监督管理局 (FDA) 批准药物的 34%。在 GPCR 的现代药理学中,根据特定疾病的需要调节受体信号传导引起了极大的兴趣。针对 GPCR 中正位位点的经典药物完全阻断内源配体的结合,从而抑制来自 GPCR 的所有重要信号。内源性配体引发的许多信号中的一些信号可能发挥重要作用,从长远来看,抑制这些信号也可能导致严重的副作用。然而,变构药物可以调节 GPCR 信号传导而不阻断内源配体结合。因此,变构药物可以维持受体的有益信号传导并防止不良副作用。在本章中,我们将讨论用变构配体解析的GPCR晶体结构、变构药物的优点以及已在临床使用或试验的变构药物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号