Current Computer-Aided Drug Design ( IF 1.5 ) Pub Date : 2020-11-30 , DOI: 10.2174/1573409915666191212152439 Meysam Shirmohammadi 1 , Esmat Mohammadinasab 1 , Zakiyeh Bayat 2
|
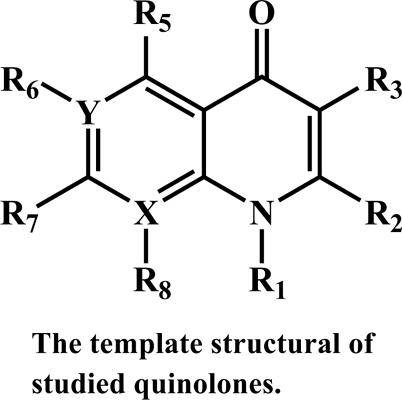
Background: In this study, we used a hierarchical approach to develop quantitative structure-activity relationship (QSAR) models for modeling physico-chemical properties of quinolone derivatives.
Objective: The relationship between some of the molecular descriptors with physic-chemical properties such as refractive index (n), polarizability (α) and HOMO-LUMO energy gap (ΔEH-L) was represented.
Materials and Methods: Quantum mechanical calculations using abinitio method at the #HF/6- 31++G** level were carried out to obtain the optimized geometry and then, the comprehensive set of molecular descriptors was computed by using the Dragon software. Genetic algorithm using multiple linear regression (GA-MLR) with backward method by SPSS software were utilized to construct QSAR models.
Results: The analytical powers of the established theoretical models were discussed using leaveone- out (LOO) cross-validation technique. A multi-parametric equation containing maximum three descriptors with suitable statistical qualities was obtained for predicting the studied properties.
Conclusion: The QSPR analysis for the prediction of the refractive index, the polarizability and the HOMO-LUMO energy gap of 40 quinolone derivatives using GA-MLR method was performed. The achieved results showed that the best model for predicting the refractive index, the polarizability and the HOMO-LUMO energy gap contains maximum three descriptors. MLR analysis, using genetic algorithms as suitable descriptors selection method showed that the three selected descriptors play a vital role in the prediction of physicochemical properties of quinolone derivatives. It can be noted that the best descriptors in the final obtained models can be used to design and screen new drugs.
中文翻译:
使用 GA-MLR 作为计算研究模拟喹诺酮衍生物的物理化学性质
背景:在本研究中,我们使用分层方法来开发定量构效关系 (QSAR) 模型,以模拟喹诺酮衍生物的理化性质。
目的:表示了一些分子描述符与物理化学性质之间的关系,例如折射率 (n)、极化率 (α) 和 HOMO-LUMO 能隙 (ΔEH-L)。
材料和方法:在#HF/6-31++G** 水平上使用abinitio 方法进行量子力学计算以获得优化的几何形状,然后使用Dragon 软件计算综合的分子描述符集。利用SPSS软件的后向法,利用多元线性回归(GA-MLR)的遗传算法构建QSAR模型。
结果:使用遗漏 (LOO) 交叉验证技术讨论了已建立的理论模型的分析能力。获得了包含最多三个具有合适统计质量的描述符的多参数方程,用于预测所研究的特性。
结论:使用 GA-MLR 方法对 40 种喹诺酮衍生物的折射率、极化率和 HOMO-LUMO 能隙进行了 QSPR 分析。取得的结果表明,预测折射率、极化率和 HOMO-LUMO 能隙的最佳模型最多包含三个描述符。MLR分析,使用遗传算法作为合适的描述符选择方法表明,三个选定的描述符在喹诺酮衍生物理化性质的预测中起着至关重要的作用。可以注意到,最终获得的模型中的最佳描述符可用于设计和筛选新药。








































 京公网安备 11010802027423号
京公网安备 11010802027423号