Surface Science ( IF 2.1 ) Pub Date : 2021-01-19 , DOI: 10.1016/j.susc.2021.121804 Víctor A. Ranea
|
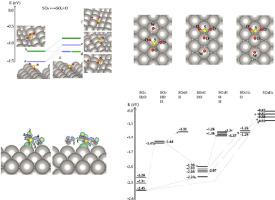
Density functional theory is used to calculate energy differences for two surface reactions on the Pt{110}() surface : SO SO + O and SO SO + O, first. Formation of SO is preferred energetically. Second, several pathways for the formation of SOH are compared from co-adsorbed SO and HO on the mentioned surface. Results reveal an energy difference (endothermic process) of 1.5 eV. However, this value decreases to 1.17 eV when the van der Waals interactions are included. Increasing the coverage of HO to four molecules per SO molecule, increases the energy difference to 1.70 eV. However, the energy difference is decreased to 1.06 eV when van der Waals interactions are taken into account in the calculations.
中文翻译:
SO的形成H 在Pt {110}()表面。DFT研究
密度泛函理论用于计算Pt {110}()表面:SO SO + O和SO 所以+ O,首先。SO的形成在能量上是优选的。二,SO形成的几种途径H 从共吸附SO进行比较 和H提到的表面上的O。结果表明,能量差(吸热过程)为1.5 eV。但是,当包括范德华相互作用时,该值降至1.17 eV。增加H的覆盖范围每个SO O到四个分子分子,将能量差增加到1.70 eV。但是,在计算中考虑了范德华相互作用时,能量差降低到1.06 eV。










































 京公网安备 11010802027423号
京公网安备 11010802027423号