Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting ligand removal energetics in thiolate-protected nanoclusters from molecular complexes
Nanoscale ( IF 5.8 ) Pub Date : 2021-1-11 , DOI: 10.1039/d0nr07839e Julia McKay 1, 2, 3, 4 , Michael J. Cowan 1, 2, 3, 4 , Cristian A. Morales-Rivera 1, 2, 3, 4 , Giannis Mpourmpakis 1, 2, 3, 4
Nanoscale ( IF 5.8 ) Pub Date : 2021-1-11 , DOI: 10.1039/d0nr07839e Julia McKay 1, 2, 3, 4 , Michael J. Cowan 1, 2, 3, 4 , Cristian A. Morales-Rivera 1, 2, 3, 4 , Giannis Mpourmpakis 1, 2, 3, 4
Affiliation
|
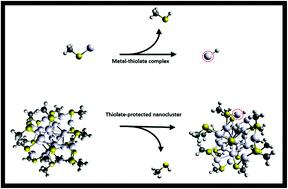
Thiolate-protected metal nanoclusters (TPNCs) have attracted great interest in the last few decades due to their high stability, atomically precise structure, and compelling physicochemical properties. Among their various applications, TPNCs exhibit excellent catalytic activity for numerous reactions; however, recent work revealed that these systems must undergo partial ligand removal in order to generate active sites. Despite the importance of ligand removal in both catalysis and stability of TPNCs, the role of ligands and metal type in the process is not well understood. Herein, we utilize Density Functional Theory to understand the energetic interplay between metal–sulfur and sulfur–ligand bond dissociation in metal–thiolate systems. We first probe 66 metal–thiolate molecular complexes across combinations of M = Ag, Au, and Cu with twenty-two different ligands (R). Our results reveal that the energetics to break the metal–sulfur and sulfur–ligand bonds are strongly correlated and can be connected across all complexes through metal atomic ionization potentials. We then extend our work to the experimentally relevant [M25(SR)18]− TPNC, revealing the same correlations at the nanocluster level. Importantly, we unify our work by introducing a simple methodology to predict TPNC ligand removal energetics solely from calculations performed on metal–ligand molecular complexes. Finally, a computational mechanistic study was performed to investigate the hydrogenation pathways for SCH3-based complexes. The energy barriers for these systems revealed, in addition to thermodynamics, that kinetics favor the break of S–R over the M–S bond in the case of the Au complex. Our computational results rationalize several experimental observations pertinent to ligand effects on TPNCs. Overall, our introduced model provides an accelerated path to predict TPNC ligand removal energies, thus aiding towards targeted design of TPNC catalysts.
中文翻译:

从分子配合物预测硫醇盐保护的纳米团簇中的配体去除能
在过去的几十年中,硫醇盐保护的金属纳米团簇(TPNC)具有很高的稳定性,原子精确的结构和令人信服的理化特性,引起了人们的极大兴趣。在其各种应用中,TPNC对许多反应均显示出优异的催化活性;但是,最近的工作表明,这些系统必须经过部分配体去除才能生成活性位点。尽管在TPNC的催化和稳定性中都需要去除配体,但对配体的作用和金属类型在该过程中的作用还知之甚少。在这里,我们利用密度泛函理论来理解金属-硫醇盐体系中金属-硫和硫-配体键解离之间的能量相互作用。我们首先在M = Ag,Au,和具有22个不同配体(R)的Cu。我们的结果表明,打破金属-硫和硫-配体键的能级具有很强的相关性,并且可以通过金属原子电离能跨所有络合物连接。然后,我们将工作扩展到与实验相关的[M25(SR) 18 ] – TPNC,揭示了纳米簇水平的相同相关性。重要的是,我们通过引入简单的方法仅通过对金属-配体分子配合物的计算来预测TPNC配体去除能,来统一我们的工作。最后,进行了计算机理研究,以研究SCH 3的加氢途径的复合体。这些系统的能量壁垒显示,除了热力学以外,在Au络合物的情况下,动力学更有利于S–R的断裂而不是M–S键的断裂。我们的计算结果使有关配体对TPNCs影响的一些实验观察合理化。总体而言,我们引入的模型为预测TPNC配体去除能提供了一条捷径,从而有助于TPNC催化剂的目标设计。
更新日期:2021-01-15
中文翻译:
从分子配合物预测硫醇盐保护的纳米团簇中的配体去除能
在过去的几十年中,硫醇盐保护的金属纳米团簇(TPNC)具有很高的稳定性,原子精确的结构和令人信服的理化特性,引起了人们的极大兴趣。在其各种应用中,TPNC对许多反应均显示出优异的催化活性;但是,最近的工作表明,这些系统必须经过部分配体去除才能生成活性位点。尽管在TPNC的催化和稳定性中都需要去除配体,但对配体的作用和金属类型在该过程中的作用还知之甚少。在这里,我们利用密度泛函理论来理解金属-硫醇盐体系中金属-硫和硫-配体键解离之间的能量相互作用。我们首先在M = Ag,Au,和具有22个不同配体(R)的Cu。我们的结果表明,打破金属-硫和硫-配体键的能级具有很强的相关性,并且可以通过金属原子电离能跨所有络合物连接。然后,我们将工作扩展到与实验相关的[M25(SR) 18 ] – TPNC,揭示了纳米簇水平的相同相关性。重要的是,我们通过引入简单的方法仅通过对金属-配体分子配合物的计算来预测TPNC配体去除能,来统一我们的工作。最后,进行了计算机理研究,以研究SCH 3的加氢途径的复合体。这些系统的能量壁垒显示,除了热力学以外,在Au络合物的情况下,动力学更有利于S–R的断裂而不是M–S键的断裂。我们的计算结果使有关配体对TPNCs影响的一些实验观察合理化。总体而言,我们引入的模型为预测TPNC配体去除能提供了一条捷径,从而有助于TPNC催化剂的目标设计。






































 京公网安备 11010802027423号
京公网安备 11010802027423号