当前位置:
X-MOL 学术
›
Phys. Status Solidi. Rapid Res. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multi‐Center Hyperbonding in Phase‐Change Materials
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-01-15 , DOI: 10.1002/pssr.202000516 Tae Hoon Lee 1 , Stephen R. Elliott 1
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-01-15 , DOI: 10.1002/pssr.202000516 Tae Hoon Lee 1 , Stephen R. Elliott 1
Affiliation
|
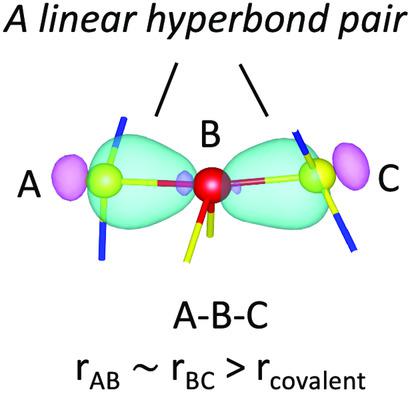
A comprehensive understanding of chemical interactions underlying the network structure of chalcogenide materials is a crucial prerequisite for comprehending their microscopic structures, physicochemical properties, and capabilities for current or potential applications. However, for many chalcogenide materials, an inherent difficulty is often present in investigating their chemical properties, due to the involvement of delocalized bonding and non‐bonding (“lone‐pair”) electrons, which requires interaction mechanisms beyond that of conventional two‐center, two‐electron covalent bonding. Herein, some recent progress in the development of new interatomic interaction models for chalcogenides is reviewed, in particular focusing on the multi‐center hyperbonding model, proposed in an effort to resolve this issue. The capability of this model in elucidating a diversity of interesting material properties of phase‐change materials (PCMs) is highlighted, including Ge2Sb2Te5 (GST). These include the propensity of high coordination numbers of constituent atoms, linear triatomic bonding geometries with short and long bonds (often ascribed to the effect of a Peierls distortion), abnormally large Born effective charges of crystalline GST, large optical contrast between amorphous and crystalline GST, ultrafast crystallization speed of amorphous GST, and the chemical origin differentiating non‐PCM from PCM chalcogenide materials. Other bonding models for these materials are also briefly discussed.
中文翻译:

相变材料中的多中心超键合
全面了解硫族化物材料网络结构下的化学相互作用是理解其微观结构,理化性质以及当前或潜在应用能力的关键前提。但是,对于许多硫族化物材料,由于其离域键合和非键合(“孤对”)电子的参与,在研究其化学性质时通常会存在固有的困难,这需要超越常规两中心电子的相互作用机理,二电子共价键。在此,回顾了硫族化物新的原子间相互作用模型开发中的一些最新进展,特别是针对解决此问题而提出的多中心超键合模型。2 Sb 2 Te 5(GST)。这些因素包括组成原子的高配位数倾向,具有短键和长键的线性三原子键几何结构(通常归因于Peierls畸变的影响),结晶GST的Born有效电荷异常大,非晶态和结晶GST之间的光学对比度大,无定形GST的超快结晶速度,以及将非PCM与PCM硫族化物材料区分开的化学起源。还简要讨论了这些材料的其他粘合模型。
更新日期:2021-03-16
中文翻译:
相变材料中的多中心超键合
全面了解硫族化物材料网络结构下的化学相互作用是理解其微观结构,理化性质以及当前或潜在应用能力的关键前提。但是,对于许多硫族化物材料,由于其离域键合和非键合(“孤对”)电子的参与,在研究其化学性质时通常会存在固有的困难,这需要超越常规两中心电子的相互作用机理,二电子共价键。在此,回顾了硫族化物新的原子间相互作用模型开发中的一些最新进展,特别是针对解决此问题而提出的多中心超键合模型。2 Sb 2 Te 5(GST)。这些因素包括组成原子的高配位数倾向,具有短键和长键的线性三原子键几何结构(通常归因于Peierls畸变的影响),结晶GST的Born有效电荷异常大,非晶态和结晶GST之间的光学对比度大,无定形GST的超快结晶速度,以及将非PCM与PCM硫族化物材料区分开的化学起源。还简要讨论了这些材料的其他粘合模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号