当前位置:
X-MOL 学术
›
J. Raman Spectrosc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Phase transition and trigger mechanism of initial reaction under pressure for benzofuroxan energetic materials: Raman spectroscopy and first‐principle calculations
Journal of Raman Spectroscopy ( IF 2.5 ) Pub Date : 2021-01-03 , DOI: 10.1002/jrs.6060 Yajing Peng 1 , Shuang Sun 1 , Jianing Meng 1 , Yuhui Liu 1 , Yunfei Song 2 , Yanqiang Yang 2
Journal of Raman Spectroscopy ( IF 2.5 ) Pub Date : 2021-01-03 , DOI: 10.1002/jrs.6060 Yajing Peng 1 , Shuang Sun 1 , Jianing Meng 1 , Yuhui Liu 1 , Yunfei Song 2 , Yanqiang Yang 2
Affiliation
|
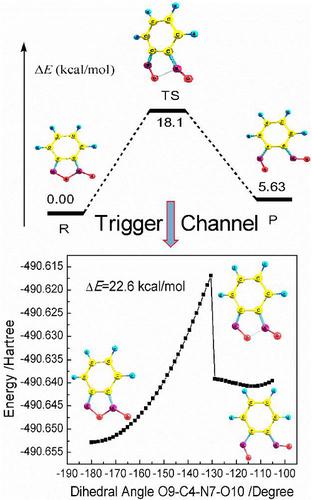
Low‐pressure (below 2 GPa) Raman spectra of benzofuroxan are investigated using a gasketed Mao–Bell‐type sapphire anvil cell and Raman spectrometer to clarify the pressure‐induced structural change and molecular vibration behavior. The first‐principle calculations are performed to compare with the experimental data and to analyze the phase transition and trigger mechanism of initial reaction. Variations of pressure‐induced Raman band width, shift, and intensity are examined. The results show that the benzofuroxan molecule will become nonplanar with increasing pressure. A phase transition occurs because of an abrupt redshift in shift–pressure relationship and reduction of the cell volume, appearing of a new vibration band above 0.13 GPa. Several active vibration modes are found, and the effects of the active mode vibrations on the initial decomposition of benzofuroxan are analyzed using the relaxed scan method for the main change in bond length, bond angle, or dihedral angle to obtain the optimal reaction channel leading to initial decomposition. The results demonstrate that the initial decomposition is the open‐loop reaction in N(O)–O position, which is originated from the increase of dihedral angle O‐C‐N(O)‐O from the out‐of‐plane torsional vibration of furoxan ring (518 cm−1). The scanning energy barrier related to dihedral angle O‐C‐N(O)‐O is about 22.6 kcal/mol, which is consistent with the calculated activation barrier (18.1 kcal/mol) of open‐loop reaction. This proves the reliability of our conclusions.
中文翻译:

苯并呋喃类高能材料在压力下的初始反应的相变和触发机制:拉曼光谱和第一性原理计算
使用衬垫的毛-贝尔型蓝宝石砧盒和拉曼光谱仪研究了苯并呋喃的低压(低于2 GPa)拉曼光谱,以阐明压力引起的结构变化和分子振动行为。进行第一性原理计算以与实验数据进行比较,并分析初始反应的相变和触发机理。检查了压力引起的拉曼带宽,位移和强度的变化。结果表明,随着压力的增加,苯并呋喃类分子将变为非平面。由于转变压力关系中的突然红移和细胞体积的减少,出现了相变,出现了高于0.13 GPa的新振动带。找到了几种主动振动模式,然后采用弛豫扫描法分析了键合长度,键角或二面角的主要变化,从而研究了主动模式振动对苯并呋喃的初始分解的影响,以获得导致初始分解的最佳反应通道。结果表明,初始分解是N(O)–O位置的开环反应,其起因于平面外扭转振动导致二面角O‐C‐N(O)‐O的增加呋喃an环(518厘米)-1)。与二面角OC-N(O)-O相关的扫描能垒约为22.6 kcal / mol,与开环反应的计算活化能垒(18.1 kcal / mol)一致。这证明了我们结论的可靠性。
更新日期:2021-01-03
中文翻译:
苯并呋喃类高能材料在压力下的初始反应的相变和触发机制:拉曼光谱和第一性原理计算
使用衬垫的毛-贝尔型蓝宝石砧盒和拉曼光谱仪研究了苯并呋喃的低压(低于2 GPa)拉曼光谱,以阐明压力引起的结构变化和分子振动行为。进行第一性原理计算以与实验数据进行比较,并分析初始反应的相变和触发机理。检查了压力引起的拉曼带宽,位移和强度的变化。结果表明,随着压力的增加,苯并呋喃类分子将变为非平面。由于转变压力关系中的突然红移和细胞体积的减少,出现了相变,出现了高于0.13 GPa的新振动带。找到了几种主动振动模式,然后采用弛豫扫描法分析了键合长度,键角或二面角的主要变化,从而研究了主动模式振动对苯并呋喃的初始分解的影响,以获得导致初始分解的最佳反应通道。结果表明,初始分解是N(O)–O位置的开环反应,其起因于平面外扭转振动导致二面角O‐C‐N(O)‐O的增加呋喃an环(518厘米)-1)。与二面角OC-N(O)-O相关的扫描能垒约为22.6 kcal / mol,与开环反应的计算活化能垒(18.1 kcal / mol)一致。这证明了我们结论的可靠性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号