Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.jmgm.2020.107830 Ndika Ngomb Simon Claude 1 , Tamafo Fouegue Aymard Didier 2 , Ateba Amana Baruch 1 , Zobo Mfomo Joseph 3 , Mbaze Meva'a Luc 1 , Bikele Mama Désiré 1
|
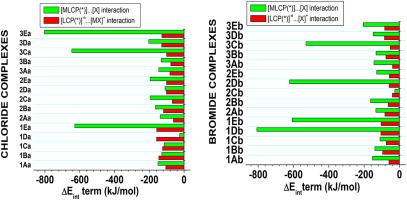
In this paper, we have explored the bonding properties of a series of mononuclear half-sandwich nd7 anticancer complexes based on N∩O dendritic scaffolds (L) using two functionals (B3LYP and BP86) with generic basis set (LanL2DZ for transition metals (as well as halogen atoms) and 6-311 + G (d,p) for others atoms. The geometry optimization of structures have led to the adoption of the piano-stool environment and the formation of kings of intermolecular hydrogen bonding: CH … X (Cl,Br) (2.619–2.954) and CH...O (2.266–2.973 Å) interaction. The metal (M)-bromine bond distances have shown to be significantly higher than metal-chlorine ones. In chloride complexes, salicylaldimine ligand-Co2+ (−3097.15 kJ/mol) and salicylaldimine ligand-Ir2+ (−3436.78 kJ/mol) interactions are stronger. Except for cobalt complexes, the interaction energies are underestimated by B3LYP functional, by contrast B3LYP HOMO-LUMO gaps obtained are highly greater. The metal ion affinity (MIA) is increasing in the order: for both halogen ligands adopted, exception for picolinate and salicylaldimine ligands. Our results also show that the metal … oxygen, metal … nitrogen and metal … halogen interactions are closed-shell interaction. From the contribution of the term in the ranges 4.06–98.61% (X = Cl) and 10.29–99.87% (X = Br), the nature of the … interaction turned out to be mainly dependent on the transition metal and halogen atom involved. The smallest back-donation observed for chloride complexes corresponding to the highest barrier to the formation of bond traduces that the fact that chloride complexes are the least reactive. In the majority of cases, larger donation is obtained compared to back-donation showing that the possibility of charge separation can be expected. For α rings of chloride complexes, B3LYP HOMA indexes are higher than those estimated by PB86 functional. Conversely, opposite results were found for their bromide counterparts.
中文翻译:
DFT / B3LYP和BP86检查基于N∩O树突状支架的单核半夹心nd 7金属药物复合物
在本文中,我们已经探索了一系列单核半夹心ND的粘结性能7个基于抗癌络合物N∩O使用两个函(B3LYP和BP86)与通用基组树突状支架(L)(对于LANL2DZ过渡金属(以及卤素原子)和其他原子的6-311 + G(d,p)。结构的几何优化使钢琴凳环境得以采用,并形成了分子间氢键之王:CH…X (Cl,Br)(2.619-2.954)和CH ... O(2.266-2.973Å)相互作用。金属(M)-溴键的距离已显着高于金属-氯键的距离。在氯化物络合物中,水杨醛亚胺配体-Co 2 +(-3097.15 kJ / mol)和水杨醛亚胺配体-Ir 2+(−3436.78 kJ / mol)相互作用更强。除钴配合物外,B3LYP官能团低估了相互作用能,相反,获得的B3LYP HOMO-LUMO间隙要大得多。金属离子亲和力(MIA)按以下顺序增加:对于采用的两个卤素配体,吡啶甲酸和水杨醛亚胺配体除外。我们的结果还表明,金属……氧,金属……氮和金属……卤素相互作用是闭壳相互作用。从的贡献 该术语的性质在4.06–98.61%(X = Cl)和10.29–99.87%(X = Br)之间 … 相互作用主要取决于所涉及的过渡金属和卤素原子。观察到的最小的氯化物络合物的背捐赠对应于最高的形成障碍键表明氯化物络合物的反应性最低。在大多数情况下,相比于反向捐赠,可以获得更大的捐赠,这表明了 可以预期电荷分离。对于氯化物配合物的α环,B3LYP HOMA指数高于PB86功能估计的那些。相反,他们的溴化物对应物却发现了相反的结果。










































 京公网安备 11010802027423号
京公网安备 11010802027423号