Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2020-12-18 , DOI: 10.1016/j.jmgm.2020.107820 L S Barbosa 1 , E Moreira 2 , A R Lopes 3 , A L A Fonseca 1 , D L Azevedo 4
|
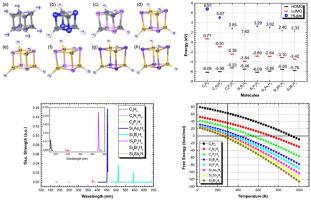
In this paper, we report structural, electronic and optical properties of cubane (C8H8) and cubanoids (cubane-like molecules) using Density Functional Theory (DFT). The cubanoids are cubanes for which Carbon atoms have been substituted by Nitrogen (N), Phosphorus (P), Boron (B), Silicon (Si), Arsenic (As), Antimony (Sb) or Bismuth (Bi) atoms. These molecules presented exceptional stability with several different symmetry point groups, being the majority Td. All calculated vibrational frequencies are positive for any studied molecules indicating that all these structures are in a stable state. The HOMO-LUMO gaps and DOS were calculated converged towards to values between 1.87 eV and 5.61 eV, actually showing promising electronic properties (Just for comparison, the cubane energy gap is 7.50 eV). The optical absorptions were also calculated for the cubanoid structure using the Time-Dependent Density Functional Theory (TD-DFT). Their dependence on the wavelength is analyzed, where five of theses structures absorb on the visible region. Finally, the extrapolation of thermodynamic properties indicates that these cubanoid could be potentially synthesized spontaneously, where four structures, the synthesis would occur for temperatures below 400 K, while for Si4Bi4H4 structure, the synthesis would occur at room temperature.
中文翻译:
古巴和古巴:DFT和TD-DFT方法的结构,光电和热力学性质
在本文中,我们使用密度泛函理论(DFT)报告了古巴(C 8 H 8)和古巴类(类古巴分子)的结构,电子和光学性质。拟南芥是碳原子已被氮(N),磷(P),硼(B),硅(Si),砷(As),锑(Sb)或铋(Bi)原子取代的古巴。这些分子在几个不同的对称点组中表现出出色的稳定性,是大多数T d。对于任何研究的分子,所有计算出的振动频率均为正值,表明所有这些结构均处于稳定状态。计算得出HOMO-LUMO间隙和DOS收敛到1.87 eV和5.61 eV之间的值,实际上显示出令人鼓舞的电子特性(为便于比较,古巴能隙为7.50 eV)。还使用时变密度泛函理论(TD-DFT)计算了蛇形结构的光吸收。分析了它们对波长的依赖性,其中五个结构在可见光区域吸收。最后,热力学性质的推断表明,这些类蛇毒菌可能是自发合成的,在四种结构中,温度低于400 K时会发生合成,而对于Si 4 Bi4 H 4结构,合成将在室温下发生。










































 京公网安备 11010802027423号
京公网安备 11010802027423号