Materials Today Advances ( IF 8.1 ) Pub Date : 2020-12-10 , DOI: 10.1016/j.mtadv.2020.100118 S. Bae , Y.-G. Kang , M. Khazaei , K. Ohno , Y.-H. Kim , M.J. Han , K.J. Chang , H. Raebiger
|
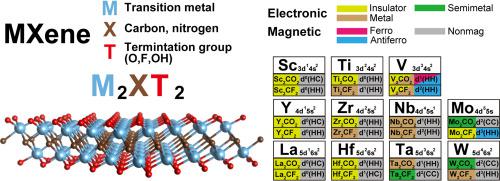
Transition metal compounds are known to be tricky for ab initio calculations mainly because of the strongly localized nature of transition metal d electrons. Nonetheless, the bulk of current theoretical studies of MXenes (transition metal carbide or nitride) relies on the density functional theory using a semilocal PBE functional, whose notorious self-interaction error grossly misrepresents electronic and magnetic properties of many well-known transition metal compounds. Although several studies have adopted Hubbard-U corrections to MXenes, the lack of systematic guidelines on how to determine the U parameters has led to a cornucopia of different results. To shed some light on the reliability of different methods (different functionals or different U parameters), we performed ab initio calculations for 22 carbide MXenes (M2CT2 with M= Sc, Y, La, Ti, Zr, Hf, V, Nb, Ta, Mo, W, and T= O, F) using density functional theory and four different methods: PBE, SCAN, HSE06, and PBE+U. In addition to trivial improvement of electronic structures of MXenes by SCAN, HSE06, and PBE+U, we found new ground-state structures for two MXenes (Hf2CF2 and Ta2CF2) and magnetic states for three MXenes (V2CO2, V2CF2, and Mo2CF2), which have not previously been reported. In addition, we found that SCAN, HSE06, and PBE+U dramatically improve the dynamical stability of V2CO2 and Mo2CF2 compared with PBE. This paper offers an overview of a broad range of MXenes with a systematic verification of various methods.
中文翻译:
碳化物MXene的电子和磁性-电子相关性的作用
众所周知,过渡金属化合物对于从头算是很棘手的,主要是因为过渡金属d电子具有很强的局部性。但是,当前对MXene(过渡金属碳化物或氮化物)的理论研究大部分依赖于使用半局部PBE官能团的密度泛函理论,其臭名昭著的自相互作用误差严重误解了许多众所周知的过渡金属化合物的电子和磁性。尽管有几项研究对MXene进行了Hubbard- U校正,但缺乏有关如何确定U的系统指导参数导致了聚宝盆的不同结果。为了阐明不同方法(不同的功能或不同的U参数)的可靠性,我们从头算了22种硬质合金MXene(M 2 CT 2,M = Sc,Y,La,Ti,Zr,Hf,V,铌,钽,钼,钨,和T = O,F),使用密度泛函理论和四种不同的方法:PBE,SCAN,HSE06,和PBE + ü。除了通过SCAN,HSE06和PBE + U改善MXene的电子结构外,我们还发现了两种MXene(Hf 2 CF 2和Ta 2 CF 2)和三个MXene(V 2 CO 2,V 2 CF 2和Mo 2 CF 2)的磁态,以前没有报道过。此外,我们发现,与PBE相比,SCAN,HSE06和PBE + U可显着提高V 2 CO 2和Mo 2 CF 2的动力稳定性。本文概述了各种MXene,并对各种方法进行了系统验证。










































 京公网安备 11010802027423号
京公网安备 11010802027423号