Chemical Physics ( IF 2.0 ) Pub Date : 2020-12-09 , DOI: 10.1016/j.chemphys.2020.111075 Diwen Liu , Luting Liang , Rongjian Sa
|
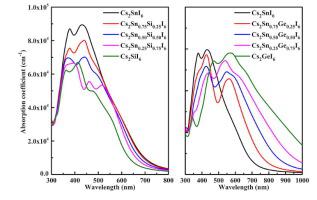
In this study, the structural stability, electronic and optical properties of all-inorganic mixed-metal double perovskites Cs2Sn1-xBxI6 (B = Si, Ge; x = 0, 0.25, 0.50, 0.75, 1) have been explored by using first-principles calculations. Our calculated results show that Cs2SnI6 shows excellent stability, while Cs2SiI6 and Cs2GeI6 are unstable. Moreover, the structural stability of Cs2SnI6 is reduced by doping Si or Ge. The predicted band gaps of the Cs2Sn1-xSixI6 systems are in the range of 1.1−1.3 eV. The band gaps of the Cs2Sn1-xGexI6 systems decrease gradually with the increasing concentration of Ge doping. Cs2SnI6 exhibits the highest optical absorption coefficient in the visible light region among the Cs2Sn1-xSixI6 systems. The Cs2Sn1-xGexI6 systems show stronger absorption coefficients in the range of 500−1000 nm compared to the pure Cs2SnI6 perovskite. Based on our calculated results, Cs2SnI6 is expected to become the most promising candidate material for perovskite solar cells.
中文翻译:
双钙钛矿Cs 2 Sn 1- x B x I 6(B = Si,Ge; x = 0,0.25,0.50,0.75,1)的结构,电子和光学性质的第一性原理计算
在这项研究中,全无机混合金属双钙钛矿Cs 2 Sn 1- x B x I 6(B = Si,Ge; x = 0,0.25,0.50,0.75,1)的结构稳定性,电子和光学性质已经通过使用第一性原理计算进行了探索。我们的计算结果表明,Cs 2 SnI 6具有出色的稳定性,而Cs 2 SiI 6和Cs 2 GeI 6不稳定。此外,通过掺杂Si或Ge降低了Cs 2 SnI 6的结构稳定性。Cs 2 Sn的预测带隙1- x Si x I 6系统的范围为1.1-1.3 eV。Cs 2 Sn 1- x Ge x I 6系统的带隙随着Ge掺杂浓度的增加而逐渐减小。在Cs 2 Sn 1- x Si x I 6系统中,Cs 2 SnI 6在可见光区域显示最高的光吸收系数。Cs 2 Sn 1- x Ge x I 6与纯Cs 2 SnI 6钙钛矿相比,该系统在500-1000 nm范围内显示出更强的吸收系数。根据我们的计算结果,Cs 2 SnI 6有望成为钙钛矿太阳能电池最有希望的候选材料。










































 京公网安备 11010802027423号
京公网安备 11010802027423号