当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Two conformational polymorphs of 4‐methylhippuric acid
Acta Crystallographica Section B ( IF 1.3 ) Pub Date : 2020-12-07 , DOI: 10.1107/s2052520620013773 Marilia Guillén , Asiloé J. Mora , Lusbely M. Belandria , Luis E. Seijas , Jeans W. Ramírez , José L. Burgos , Luis Rincón , Gerzon E. Delgado
Acta Crystallographica Section B ( IF 1.3 ) Pub Date : 2020-12-07 , DOI: 10.1107/s2052520620013773 Marilia Guillén , Asiloé J. Mora , Lusbely M. Belandria , Luis E. Seijas , Jeans W. Ramírez , José L. Burgos , Luis Rincón , Gerzon E. Delgado
|
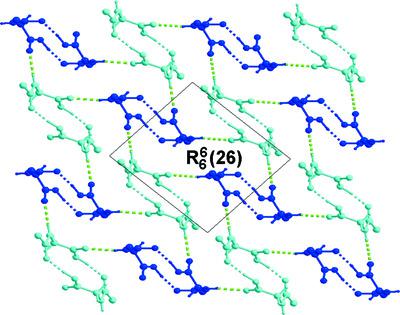
4‐Methylhippuric acid {systematic name: 2‐[(4‐methylbenzoyl)amino]ethanoic acid}, a p‐xylene excreted metabolite with a backbone containing three rotatable bonds (R‐bonds), is likely to produce more than one stable molecular structure in the solid state. In this work, we prepared polymorph I by slow solvent evaporation (plates with Z′ = 1) and polymorph II by mechanical grinding (plates with Z′ = 2). Potential energy surface (PES) analysis, rotating the molecule about the C—C—N—C torsion angle, shows four conformational energy basins. The second basin, with torsion angles near −73°, agree with the conformations adopted by polymorph I and molecules A of polymorph II, and the third basin at 57° matched molecules B of polymorph II. The energy barrier between these basins is 27.5 kJ mol−1. Superposition of the molecules of polymorphs I and II rendered a maximum r.m.s. deviation of 0.398 Å. Polymorphs I and II are therefore true conformational polymorphs. The crystal packing of polymorph I consists of C(5) chains linked by N—H…O interactions along the a axis and C(7) chains linked by O—H…O interactions along the b axis. In polymorph II, two molecules (A with A or B with B) are connected by two acid–amide O—H…O interactions rendering R22(14) centrosymmetric dimers. These dimers alternate to pile up along the b axis linked by N—H…O interactions. A Hirshfeld surface analysis localized weaker noncovalent interactions, C—H…O and C—H…π, with contact distances close to the sum of the van der Waals radii. Electron density at a local level using the Quantum Theory of Atoms in Molecules (QTAIM) and the Electron Localization Function (ELF), or a semi‐local level using noncovalent interactions, was used to rank interactions. Strong closed shell interactions in classical O—H…O and N—H…O hydrogen bonds have electron density highly localized on bond critical points. Weaker delocalized electron density is seen around the p‐methylphenyl rings associated with dispersive C—H…π and H…H interactions.
中文翻译:

4-甲基马尿酸的两个构象多晶型物
4-甲基马尿酸{系统名称:2-[((4-甲基苯甲酰基)氨基]乙酸},一种对二甲苯排出的代谢产物,其骨架含有三个可旋转键(R-键),可能会产生多个稳定分子固态结构。在这项工作中,我们通过缓慢的溶剂蒸发(Z '= 1的板)制备了多晶型物I,并通过机械研磨(Z '= 2的板)制备了多晶型物II。势能面(PES)分析使分子绕C–N–C扭转角旋转,显示了四个构象能盆。第二个盆地的扭转角接近-73°,与多晶型物I和分子A所采用的构型一致多晶型物II的分子,并且第三个盆在57°与多晶型物II的分子B匹配。这些盆地之间的能垒为27.5 kJ mol -1。多晶型物I和II分子的叠加使最大均方根偏差为0.398Å。因此,多晶型物I和II是真正的构象多晶型物。多晶型物I的晶体堆积由沿a轴通过NH…O相互作用连接的C(5)链 和沿b轴通过OH…O相互作用连接的C(7)链组成。在多晶型物II中,两个分子(A与A或B与B)通过两个酸-酰胺O-H…O相互作用连接,从而形成R 2 2(14)中心对称二聚体。这些二聚体交替沿b堆积通过N–H…O相互作用链接的轴。Hirshfeld表面分析确定了较弱的非共价相互作用,即CH…O和CH…π,接触距离接近范德华半径之和。使用分子中的原子量子理论(QTAIM)和电子本地化函数(ELF)在局部水平上的电子密度,或使用非共价相互作用的半局部水平上的电子密度来对相互作用进行排名。经典的O-H…O和N-H…O氢键中的强闭合壳相互作用使电子密度高度集中在键的临界点上。在与分散的CH-π和H-H相互作用相关的对-甲基苯基环周围发现较弱的离域电子密度。
更新日期:2020-12-07
中文翻译:
4-甲基马尿酸的两个构象多晶型物
4-甲基马尿酸{系统名称:2-[((4-甲基苯甲酰基)氨基]乙酸},一种对二甲苯排出的代谢产物,其骨架含有三个可旋转键(R-键),可能会产生多个稳定分子固态结构。在这项工作中,我们通过缓慢的溶剂蒸发(Z '= 1的板)制备了多晶型物I,并通过机械研磨(Z '= 2的板)制备了多晶型物II。势能面(PES)分析使分子绕C–N–C扭转角旋转,显示了四个构象能盆。第二个盆地的扭转角接近-73°,与多晶型物I和分子A所采用的构型一致多晶型物II的分子,并且第三个盆在57°与多晶型物II的分子B匹配。这些盆地之间的能垒为27.5 kJ mol -1。多晶型物I和II分子的叠加使最大均方根偏差为0.398Å。因此,多晶型物I和II是真正的构象多晶型物。多晶型物I的晶体堆积由沿a轴通过NH…O相互作用连接的C(5)链 和沿b轴通过OH…O相互作用连接的C(7)链组成。在多晶型物II中,两个分子(A与A或B与B)通过两个酸-酰胺O-H…O相互作用连接,从而形成R 2 2(14)中心对称二聚体。这些二聚体交替沿b堆积通过N–H…O相互作用链接的轴。Hirshfeld表面分析确定了较弱的非共价相互作用,即CH…O和CH…π,接触距离接近范德华半径之和。使用分子中的原子量子理论(QTAIM)和电子本地化函数(ELF)在局部水平上的电子密度,或使用非共价相互作用的半局部水平上的电子密度来对相互作用进行排名。经典的O-H…O和N-H…O氢键中的强闭合壳相互作用使电子密度高度集中在键的临界点上。在与分散的CH-π和H-H相互作用相关的对-甲基苯基环周围发现较弱的离域电子密度。









































 京公网安备 11010802027423号
京公网安备 11010802027423号