European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2020-12-04 , DOI: 10.1016/j.ejmech.2020.113081 Rawan M. Sbenati , Seyed-Omar Zaraei , Mohammed I. El-Gamal , Hanan S. Anbar , Hamadeh Tarazi , Malaka M. Zoghbor , Najma A. Mohamood , Mahta M. Khakpour , Dana M. Zaher , Hany A. Omar , Nour N. Alach , Mahmoud K. Shehata , Randa El-Gamal
|
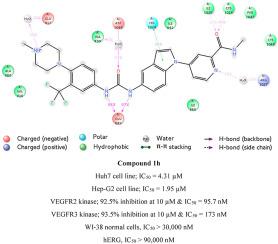
Sorafenib is one of the clinically used anticancer agents that inhibits several kinases. In this study, novel indole-based rigid analogues of sorafenib were designed and synthesized in order to enhance kinase selectivity and hence minimize the side effects associated with its use. The target compounds possess different linkers; urea, amide, sulfonamide, or thiourea, in addition to different terminal aryl moieties attached to the linker in order to investigate their impact on biological activity. They were tested against Hep3B, Huh7, and Hep-G2 hepatocellular carcinoma (HCC) cell lines to study their potency. Among all the tested target derivatives, compound 1h exerted superior antiproliferative potency against all the three tested HCC cell lines compared to sorafenib. Based on these preliminary results, compound 1h was selected for further biological and in silico investigations. Up to 30 μM, compound 1h did not inhibit 50% of the proliferation of WI-38 normal cells, which indicated promising selectivity against HCC cells than normal cells. In addition, compound 1h exerted superior kinase selectivity than sorafenib. It is selective for VEGFR2 and VEGFR3 angiogenesis-related kinases, while sorafenib is a multikinase inhibitor. Superior kinase selectivity of compound 1h to sorafenib can be attributed to its conformationally-restricted indole nucleus and the bulky N-methylpiperazinyl moiety. Western blotting was carried out and confirmed the ability of compound 1h to inhibit VEGFR2 kinase inside Hep-G2 HCC cells in a dose-dependent pattern. Compound 1h induces apoptosis and necrosis in Hep-G2 cell line, as shown by caspase-3/7 and lactate dehydrogenase (LDH) release assays, respectively. Moreover, compound 1h is rather safe against hERG. Thus, we could achieve a more selective kinase inhibitor than sorafenib with retained or even better antiproliferative potency against HCC cell lines. Furthermore, molecular docking and dynamic simulation studies were carried out to investigate its binding mode with VEGFR2 kinase. The molecule has a unique orientation upon binding with the kinase.
中文翻译:
设计,合成,生物学评估和建模研究索拉非尼新型构象受限的类似物,作为针对肝癌细胞的选择性激酶抑制性抗增殖药
索拉非尼是临床上使用的抑制几种激酶的抗癌药之一。在这项研究中,索拉非尼的新型基于吲哚的刚性类似物被设计和合成,以增强激酶的选择性,从而最小化与其使用相关的副作用。目标化合物具有不同的接头。尿素,酰胺,磺酰胺或硫脲,以及连接在接头上的不同末端芳基部分,以研究其对生物活性的影响。他们针对Hep3B,Huh7和Hep-G2肝细胞癌(HCC)细胞系进行了测试,以研究其功效。在所有测试的目标衍生物中,化合物1h与索拉非尼相比,对所有三种测试的HCC细胞系均表现出优异的抗增殖能力。根据这些初步结果,选择了化合物1h用于进一步的生物学和计算机分析研究。高达30μM,化合物1h不会抑制WI-38正常细胞50%的增殖,这表明对HCC细胞的选择性比正常细胞有希望。另外,化合物1h比索拉非尼具有更好的激酶选择性。它对VEGFR2和VEGFR3血管生成相关激酶具有选择性,而索拉非尼是一种多激酶抑制剂。化合物1h对索拉非尼的优异激酶选择性可归因于其构象受限的吲哚核和大体积N-甲基哌嗪基部分。进行了蛋白质印迹,并证实了化合物1h以剂量依赖的方式抑制Hep-G2 HCC细胞内部的VEGFR2激酶的能力。化合物1h在Hep-G2细胞系中诱导凋亡和坏死,分别由caspase-3 / 7和乳酸脱氢酶(LDH)释放试验表明。此外,化合物1h对hERG相当安全。因此,我们可以获得比索拉非尼更具选择性的激酶抑制剂,并具有对HCC细胞系的保留或什至更好的抗增殖能力。此外,进行了分子对接和动态模拟研究以研究其与VEGFR2激酶的结合模式。该分子与激酶结合后具有独特的方向。








































 京公网安备 11010802027423号
京公网安备 11010802027423号