当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Improving the Description of Interlayer Bonding in TiS2 by Density Functional Theory
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jpcc.0c09460 Matteo Ricci 1 , Alberto Ambrosetti 1 , Pier Luigi Silvestrelli 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jpcc.0c09460 Matteo Ricci 1 , Alberto Ambrosetti 1 , Pier Luigi Silvestrelli 1
Affiliation
|
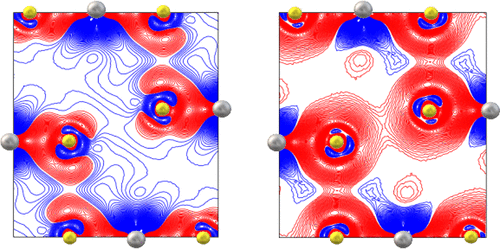
We investigate energetic and electronic properties of TiS2, an archetypal van der Waals (vdW) material, from first principles, in the framework of density functional theory (DFT). In this system a recent experimental study showed a puzzling discrepancy between the distribution of the electron density in the interlayer region obtained by X-ray diffraction data and that computed by DFT, even adopting DFT functionals that should properly include vdW effects. Such a discrepancy could indicate a partial failure of state-of-the-art DFT approaches in describing the weak interlayer interactions of TiS2 and, possibly, of similar systems too. In order to shed light on this issue, we have carried out simulations based on different DFT functionals, basically confirming the mentioned discrepancy with the experimental findings. Subsequently, we have tried to reproduce the experimental interlayer electronic density deformation by changing the parameters characterizing the rVV10 DFT functional (in such a way to artificially modify the strength of the vdW interactions at short or long-range) and also by adopting a modified pseudopotential for sulfur atoms, involving d orbitals. The latter approach turns out to be particularly promising. In fact, using this novel, more flexible pseudopotential, we obtain not only an electronic density deformation closer to the experimental profile, but also a better estimate of the interlayer binding energy. Interestingly, this improvement in the theoretical DFT description is not limited to TiS2 but also applies to other similar layered systems involving S atoms, such as TaS2, HfS2, and MoS2.
中文翻译:

用密度泛函理论改进TiS 2中层间键合的描述
我们在密度泛函理论(DFT)的框架内,从第一原理出发,研究了原型范德华(vdW)材料TiS 2的能量和电子性质。在该系统中,最近的一项实验研究表明,即使采用应该适当包括vdW效应的DFT功能,通过X射线衍射数据获得的夹层区域中电子密度的分布与通过DFT计算得出的电子密度之间的差异也令人费解。这种差异可能表明,现有的DFT方法在描述TiS 2的弱层间相互作用方面存在部分失败可能还有类似的系统。为了阐明这一问题,我们基于不同的DFT功能进行了仿真,基本上从实验结果上证实了上述差异。随后,我们尝试通过更改表征rVV10 DFT功能的参数(以这种方式人为地修改短距离或长距离的vdW相互作用的强度)并采用修改的伪势来重现实验性的层间电子密度变形。硫原子,涉及d轨道。事实证明后一种方法特别有前途。实际上,使用这种新颖,更灵活的pseudo势,我们不仅获得了更接近实验分布的电子密度变形,而且还获得了更好的层间结合能估计值。2,但也适用于涉及S原子,如TAS其它类似的层状系统2,HFS 2,和MoS 2。
更新日期:2020-12-17
中文翻译:
用密度泛函理论改进TiS 2中层间键合的描述
我们在密度泛函理论(DFT)的框架内,从第一原理出发,研究了原型范德华(vdW)材料TiS 2的能量和电子性质。在该系统中,最近的一项实验研究表明,即使采用应该适当包括vdW效应的DFT功能,通过X射线衍射数据获得的夹层区域中电子密度的分布与通过DFT计算得出的电子密度之间的差异也令人费解。这种差异可能表明,现有的DFT方法在描述TiS 2的弱层间相互作用方面存在部分失败可能还有类似的系统。为了阐明这一问题,我们基于不同的DFT功能进行了仿真,基本上从实验结果上证实了上述差异。随后,我们尝试通过更改表征rVV10 DFT功能的参数(以这种方式人为地修改短距离或长距离的vdW相互作用的强度)并采用修改的伪势来重现实验性的层间电子密度变形。硫原子,涉及d轨道。事实证明后一种方法特别有前途。实际上,使用这种新颖,更灵活的pseudo势,我们不仅获得了更接近实验分布的电子密度变形,而且还获得了更好的层间结合能估计值。2,但也适用于涉及S原子,如TAS其它类似的层状系统2,HFS 2,和MoS 2。









































 京公网安备 11010802027423号
京公网安备 11010802027423号