当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of Perturbative Vibronic Correction for Weak Fluorescence in Thermally Activated Delayed Fluorescence Systems
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-27 , DOI: 10.1021/acs.jpca.0c09439 Soo Wan Park 1 , Joong Hwan Yang 2 , Hyongjong Choi 2 , Young Min Rhee 1 , Dongwook Kim 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-27 , DOI: 10.1021/acs.jpca.0c09439 Soo Wan Park 1 , Joong Hwan Yang 2 , Hyongjong Choi 2 , Young Min Rhee 1 , Dongwook Kim 3
Affiliation
|
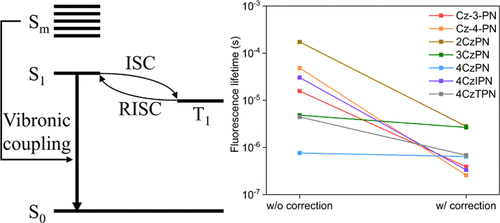
Minimizing the energy difference between the lowest singlet (S1) and the lowest triplet states, ΔEST, is the main strategy to design thermally activated delayed fluorescence (TADF) molecules, and spatially separating the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is the general method in the design. However, such a separation also tends to reduce the oscillator strength of the S1 state. In real systems, vibrations change the S1 oscillator strength, and thus one needs to consider the vibronic coupling toward searching for TADF candidate molecules. Here, we evaluate the importance of vibronic coupling by including the first-order perturbative correction to the transition dipole moments of carbazolyl-phthalonitrile derivatives. Indeed, some molecules display large enhancements in their oscillator strengths, with their fluorescence lifetimes reduced by 2 orders of magnitude. The twisting mode between the carbazole groups and phthalonitrile is the most important mode in inducing the perturbations. Thus, performing the perturbative correction is crucial in attaining more reliable predictions on their fluorescence propensities. We also observe that some other molecules, whose zeroth-order predicted fluorescence rates are much slower than the actual experimental data, are affected little by the same first-order correction. For these molecules, we deduce that the geometry-dependent excited-state switching kicks in. Our results demonstrate the significance of vibronic coupling in TADF molecules and the importance of adopting correction schemes as the guidelines for screening of useful TADF molecules.
中文翻译:

微扰振动校正对热激活延迟荧光系统中弱荧光的影响
最小化最低单重态(S 1)和最低三重态之间的能量差ΔE ST是设计热激活延迟荧光(TADF)分子并在空间上将最高占据分子轨道(HOMO)与高能分子分开的主要策略。最小的未占用分子轨道(LUMO)是设计中的常规方法。然而,这种分离也趋于降低S 1状态的振荡器强度。在实际系统中,振动会改变S 1振动强度,因此需要考虑振动耦合以寻找TADF候选分子。在这里,我们通过对咔唑基-邻苯二甲腈衍生物的跃迁偶极矩进行一阶微扰校正来评估振动耦合的重要性。实际上,某些分子的振荡器强度显示出极大的增强,其荧光寿命降低了2个数量级。咔唑基和邻苯二甲腈之间的加捻模式是引起扰动的最重要模式。因此,执行扰动校正对于获得对其荧光倾向的更可靠预测至关重要。我们还观察到其他一些分子,其零阶预测荧光速率比实际实验数据要慢得多,受到相同的一阶校正的影响很小。对于这些分子,我们推断出了依赖于几何形状的激发态转换。我们的结果证明了TADF分子中电子耦合的重要性,以及采用校正方案作为筛选有用TADF分子的指南的重要性。
更新日期:2020-12-10
中文翻译:
微扰振动校正对热激活延迟荧光系统中弱荧光的影响
最小化最低单重态(S 1)和最低三重态之间的能量差ΔE ST是设计热激活延迟荧光(TADF)分子并在空间上将最高占据分子轨道(HOMO)与高能分子分开的主要策略。最小的未占用分子轨道(LUMO)是设计中的常规方法。然而,这种分离也趋于降低S 1状态的振荡器强度。在实际系统中,振动会改变S 1振动强度,因此需要考虑振动耦合以寻找TADF候选分子。在这里,我们通过对咔唑基-邻苯二甲腈衍生物的跃迁偶极矩进行一阶微扰校正来评估振动耦合的重要性。实际上,某些分子的振荡器强度显示出极大的增强,其荧光寿命降低了2个数量级。咔唑基和邻苯二甲腈之间的加捻模式是引起扰动的最重要模式。因此,执行扰动校正对于获得对其荧光倾向的更可靠预测至关重要。我们还观察到其他一些分子,其零阶预测荧光速率比实际实验数据要慢得多,受到相同的一阶校正的影响很小。对于这些分子,我们推断出了依赖于几何形状的激发态转换。我们的结果证明了TADF分子中电子耦合的重要性,以及采用校正方案作为筛选有用TADF分子的指南的重要性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号