当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Generalized unitary coupled cluster excitations for multireference molecular states optimized by the variational quantum eigensolver
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-11-27 , DOI: 10.1002/qua.26352 Gabriel Greene‐Diniz 1 , David Muñoz Ramo 1
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-11-27 , DOI: 10.1002/qua.26352 Gabriel Greene‐Diniz 1 , David Muñoz Ramo 1
Affiliation
|
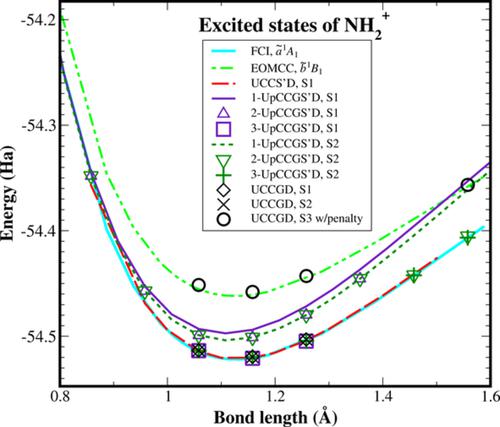
The variational quantum eigensolver (VQE) algorithm, designed to calculate the energy of molecular ground states on near-term quantum computers, requires specification of symmetries that describe the system, e.g. spin state and number of electrons. This opens the possibility of using VQE to obtain excited states as the lowest energy solutions of a given set of symmetries. In this paper, the performances of various unitary coupled cluster (UCC) ans\"atze applied to VQE calculations on excited states are investigated, using quantum circuits designed to represent single reference and multireference wavefunctions to calculate energy curves with respect to variations in the molecular geometry. These ans\"atze include standard UCCSD, as well as modified versions of UCCGSD and k-UpCCGSD which are engineered to tackle excited states without undesired spin symmetry cross-over to lower states during VQE optimization. These studies are carried out on a range of systems including H$_2$, H$_3$, H$_4$, NH, and OH$^{+}$, CH$_2$, NH$^{+}_{2}$, covering examples of spin singlet, doublet and triplet molecular ground states with single and multireference excited states. In most cases, our calculations are in excellent agreement with results from full configuration interaction calculations on classical machines, thus showing that the VQE algorithm is capable of calculating the lowest excited state at a certain symmetry, including multireference closed and open shell states, by setting appropriate restrictions on the excitations considered in the cluster operator, and appropriate constraints in the qubit register encoding the starting mean field state.
中文翻译:

变分量子本征求解器优化的多参考分子态的广义幺正耦合簇激发
变分量子本征求解器 (VQE) 算法旨在计算近期量子计算机上分子基态的能量,需要指定描述系统的对称性,例如自旋态和电子数。这开启了使用 VQE 获得激发态作为给定对称组的最低能量解的可能性。在本文中,研究了应用于激发态 VQE 计算的各种酉耦合簇 (UCC) 和“atze”的性能,使用设计用于表示单参考和多参考波函数的量子电路来计算关于分子变化的能量曲线几何形状。这些答案包括标准 UCCSD,以及 UCCGSD 和 k-UpCCGSD 的修改版本,它们旨在解决激发态,而不会在 VQE 优化过程中出现不希望的自旋对称性交叉到较低态。这些研究在一系列系统上进行,包括 H$_2$、H$_3$、H$_4$、NH 和 OH$^{+}$、CH$_2$、NH$^{+}_{ 2}$,涵盖了具有单参考和多参考激发态的自旋单线态、双线态和三线态分子基态的例子。在大多数情况下,我们的计算与经典机器上全配置相互作用计算的结果非常一致,从而表明 VQE 算法能够计算特定对称性下的最低激发态,包括多参考闭壳态和开壳态,通过设置对集群算子中考虑的激励的适当限制,
更新日期:2020-11-27
中文翻译:
变分量子本征求解器优化的多参考分子态的广义幺正耦合簇激发
变分量子本征求解器 (VQE) 算法旨在计算近期量子计算机上分子基态的能量,需要指定描述系统的对称性,例如自旋态和电子数。这开启了使用 VQE 获得激发态作为给定对称组的最低能量解的可能性。在本文中,研究了应用于激发态 VQE 计算的各种酉耦合簇 (UCC) 和“atze”的性能,使用设计用于表示单参考和多参考波函数的量子电路来计算关于分子变化的能量曲线几何形状。这些答案包括标准 UCCSD,以及 UCCGSD 和 k-UpCCGSD 的修改版本,它们旨在解决激发态,而不会在 VQE 优化过程中出现不希望的自旋对称性交叉到较低态。这些研究在一系列系统上进行,包括 H$_2$、H$_3$、H$_4$、NH 和 OH$^{+}$、CH$_2$、NH$^{+}_{ 2}$,涵盖了具有单参考和多参考激发态的自旋单线态、双线态和三线态分子基态的例子。在大多数情况下,我们的计算与经典机器上全配置相互作用计算的结果非常一致,从而表明 VQE 算法能够计算特定对称性下的最低激发态,包括多参考闭壳态和开壳态,通过设置对集群算子中考虑的激励的适当限制,









































 京公网安备 11010802027423号
京公网安备 11010802027423号