Chemical Physics Letters ( IF 2.8 ) Pub Date : 2020-11-27 , DOI: 10.1016/j.cplett.2020.138222 Takashi Ikeda
|
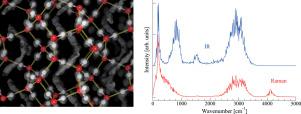
The crystal structure proposed experimentally for C0 phase of H2 hydrate has been assessed by first-principles path-integral based molecular dynamics simulations. Our constant pressure and temperature simulations in the NPT ensemble suggest that H2 hydrate in C0 phase has a hexagonal unit cell in agreement with experiments. The fractional coordinates of H2O molecules also agree well with the experimental ones, supporting the presence of interpenetrating spiral chains of H-bonded water molecules. Limitations in the linear response theory used to compute the Raman spectra allow at least for a qualitative agreement with the observed experimental ones.
中文翻译:
基于第一原理路径积分的水合氢在C 0相中的分子动力学模拟
通过第一性原理基于路径积分的分子动力学模拟,评估了H 2水合物C 0相实验提出的晶体结构。我们在NPT集合中进行的恒定压力和温度模拟表明,与实验一致,C 0相中的H 2水合物具有六角形晶胞。H 2 O分子的分数坐标也与实验结果非常吻合,支持了H键水分子相互渗透的螺旋链的存在。用于计算拉曼光谱的线性响应理论的局限性至少允许与观察到的实验定性一致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号