Chemical Physics ( IF 2.3 ) Pub Date : 2020-11-26 , DOI: 10.1016/j.chemphys.2020.111055 Qiman Liu , Yunhu Hu , Xiaoyan Zhang , Fengwu Wang , Longjiu Cheng
|
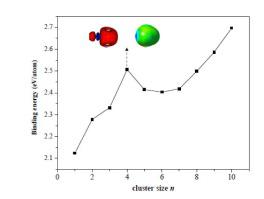
The Hume-Rothery alloy phases of the main-group elements are rather rare, of which the phase stability mechanisms are decided by the electronic effects. The intermetallic compound Be-Pt alloy is a member of this family, but structural and electronic characters of them at nanometer scale still remain unclear. In this work, the geometric structures, stabilities and electronic properties of BenPt clusters (n = 1-10) are first investigated using the method combining the genetic algorithm with density function theory (DFT). Benchmark calculations indicate that the PBE-D3 functional is reliable in predicting the energetic sequences of four isomers of Be4Pt cluster compared to the high-level coupled cluster method. In general, the most structures of the alloy clusters can be obtained by replacing one beryllium atom in the pure beryllium (Bem, m = 2-11) clusters with a Pt atom. We found that the Be4Pt and Be10Pt clusters are two very stable 8e/20e superatoms in this series, respectively, where the electronic structure of Pt atom is d10. Analyses of electrostatic potential surfaces show that these two clusters have significant σ-holes with most positive potential regions, which can be seen as binding sites with Lewis bases. When a CO molecule is adsorbed on the superatom clusters, the C=O stretching frequency has a large red-shift and the bond length is slightly elongated, due to the electrons of the clusters transfer to the anti-bond π orbital of CO. This work gives inferences for further understanding structural characters of the binary alloy from the nanoscale view.
中文翻译:
富Be纳米合金的结构,电子和吸附特性:Be n Pt(n = 1-10)簇
主族元素的休姆-拉氏合金相很少见,其相稳定机制由电子效应决定。金属间化合物Be-Pt合金是该族的一员,但是它们在纳米级的结构和电子特性仍然不清楚。在这项工作中,首先使用遗传算法和密度泛函理论(DFT)相结合的方法研究了Be n Pt团簇(n = 1-10)的几何结构,稳定性和电子性质。基准计算表明,PBE-D3官能团在预测Be 4的四个异构体的能量序列方面是可靠的Pt群集与高级耦合群集方法相比。通常,通过用Pt原子替换纯铍(Be m,m = 2-11)簇中的一个铍原子,可以获得大多数的合金簇结构。我们发现Be 4 Pt和Be 10 Pt团簇分别是该系列中的两个非常稳定的8e / 20e超原子,其中Pt原子的电子结构为d 10。静电势表面的分析表明,这两个簇具有显着的σ孔,具有最大的正电势区域,可以视为与Lewis碱的结合位点。当CO分子吸附在超原子团簇上时,由于团簇的电子转移到CO的反键π轨道上,C = O的拉伸频率具有较大的红移并且键长略微延长。这项工作为从纳米尺度进一步理解二元合金的结构特征提供了推论。


























 京公网安备 11010802027423号
京公网安备 11010802027423号