当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies
Chemical Science ( IF 8.4 ) Pub Date : 2020-11-05 , DOI: 10.1039/d0sc04896h Kjell Jorner 1 , Tore Brinck 2 , Per-Ola Norrby 3 , David Buttar 1
Chemical Science ( IF 8.4 ) Pub Date : 2020-11-05 , DOI: 10.1039/d0sc04896h Kjell Jorner 1 , Tore Brinck 2 , Per-Ola Norrby 3 , David Buttar 1
Affiliation
|
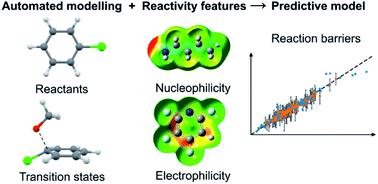
Accurate prediction of chemical reactions in solution is challenging for current state-of-the-art approaches based on transition state modelling with density functional theory. Models based on machine learning have emerged as a promising alternative to address these problems, but these models currently lack the precision to give crucial information on the magnitude of barrier heights, influence of solvents and catalysts and extent of regio- and chemoselectivity. Here, we construct hybrid models which combine the traditional transition state modelling and machine learning to accurately predict reaction barriers. We train a Gaussian Process Regression model to reproduce high-quality experimental kinetic data for the nucleophilic aromatic substitution reaction and use it to predict barriers with a mean absolute error of 0.77 kcal mol−1 for an external test set. The model was further validated on regio- and chemoselectivity prediction on patent reaction data and achieved a competitive top-1 accuracy of 86%, despite not being trained explicitly for this task. Importantly, the model gives error bars for its predictions that can be used for risk assessment by the end user. Hybrid models emerge as the preferred alternative for accurate reaction prediction in the very common low-data situation where only 100–150 rate constants are available for a reaction class. With recent advances in deep learning for quickly predicting barriers and transition state geometries from density functional theory, we envision that hybrid models will soon become a standard alternative to complement current machine learning approaches based on ground-state physical organic descriptors or structural information such as molecular graphs or fingerprints.
中文翻译:

机器学习与机械建模相结合,可准确预测实验活化能
对于当前基于密度泛函理论的过渡态建模的最新方法而言,准确预测溶液中的化学反应具有挑战性。基于机器学习的模型已成为解决这些问题的有希望的替代方案,但这些模型目前缺乏精确度,无法提供有关势垒高度大小、溶剂和催化剂的影响以及区域和化学选择性程度的关键信息。在这里,我们构建了结合传统过渡态建模和机器学习的混合模型,以准确预测反应障碍。我们训练了一个高斯过程回归模型来重现亲核芳族取代反应的高质量实验动力学数据,并用它来预测平均绝对误差为 0.77 kcal mol 的势垒-1对于外部测试集。该模型在专利反应数据的区域和化学选择性预测方面得到了进一步验证,并实现了 86% 的竞争性 top-1 准确度,尽管没有针对该任务进行明确的训练。重要的是,该模型为其预测提供了误差线,最终用户可将其用于风险评估。在非常常见的低数据情况下,混合模型成为准确反应预测的首选替代方案,其中一个反应类别只有 100-150 个速率常数可用。随着深度学习在从密度泛函理论中快速预测障碍和过渡态几何的最新进展,
更新日期:2020-11-27
中文翻译:
机器学习与机械建模相结合,可准确预测实验活化能
对于当前基于密度泛函理论的过渡态建模的最新方法而言,准确预测溶液中的化学反应具有挑战性。基于机器学习的模型已成为解决这些问题的有希望的替代方案,但这些模型目前缺乏精确度,无法提供有关势垒高度大小、溶剂和催化剂的影响以及区域和化学选择性程度的关键信息。在这里,我们构建了结合传统过渡态建模和机器学习的混合模型,以准确预测反应障碍。我们训练了一个高斯过程回归模型来重现亲核芳族取代反应的高质量实验动力学数据,并用它来预测平均绝对误差为 0.77 kcal mol 的势垒-1对于外部测试集。该模型在专利反应数据的区域和化学选择性预测方面得到了进一步验证,并实现了 86% 的竞争性 top-1 准确度,尽管没有针对该任务进行明确的训练。重要的是,该模型为其预测提供了误差线,最终用户可将其用于风险评估。在非常常见的低数据情况下,混合模型成为准确反应预测的首选替代方案,其中一个反应类别只有 100-150 个速率常数可用。随着深度学习在从密度泛函理论中快速预测障碍和过渡态几何的最新进展,


























 京公网安备 11010802027423号
京公网安备 11010802027423号