当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Neural Network Based Quasi-diabatic Representation for S0 and S1 States of Formaldehyde
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-24 , DOI: 10.1021/acs.jpca.0c08948 Yafu Guan 1 , Changjian Xie 2 , Hua Guo 3 , David R. Yarkony 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-24 , DOI: 10.1021/acs.jpca.0c08948 Yafu Guan 1 , Changjian Xie 2 , Hua Guo 3 , David R. Yarkony 1
Affiliation
|
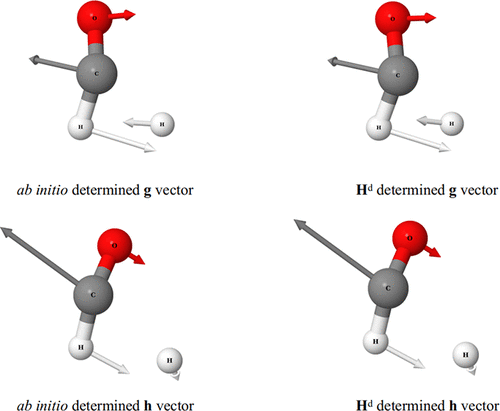
A neural network based quasi-diabatic potential energy matrix Hd that describes the photodissociation of formaldehyde involving the two lowest singlet states S0 and S1 is constructed. It has strict complete nuclear permutation inversion symmetry encoded and can reproduce high level ab initio electronic structure data, including energies, energy gradients, and derivative couplings, with excellent accuracy. It has been fully saturated in the configuration space to cover all possible reaction pathways with a trajectory-guided point sampling approach. This Hd will not only enable the accurate full-dimensional dynamic simulations of the photodissociation of formaldehyde involving S0 and S1 but also provide a crucial ingredient for incorporating spin–orbit couplings into a diabatic framework, thus ultimately enabling the study of both internal conversion and intersystem crossing in formaldehyde on the same footing.
中文翻译:

基于神经网络的甲醛S 0和S 1态的绝热表示
构造了基于神经网络的准绝热势能矩阵H d,该矩阵描述了涉及两个最低单重态S 0和S 1的甲醛的光解离。它具有严格完整的核置换反演对称性编码,并且可以以极高的精度重现高级的从头算起的电子结构数据,包括能量,能量梯度和微分耦合。它已经在配置空间中完全饱和,可以通过轨迹引导点采样方法覆盖所有可能的反应路径。此H d不仅可以对涉及S 0的甲醛进行光解反应进行准确的全尺寸动态模拟和S 1,也为将自旋-轨道偶合纳入非绝热骨架提供了至关重要的成分,因此最终使人们能够在同一基础上研究甲醛的内部转化和系统间交叉。
更新日期:2020-12-10
中文翻译:
基于神经网络的甲醛S 0和S 1态的绝热表示
构造了基于神经网络的准绝热势能矩阵H d,该矩阵描述了涉及两个最低单重态S 0和S 1的甲醛的光解离。它具有严格完整的核置换反演对称性编码,并且可以以极高的精度重现高级的从头算起的电子结构数据,包括能量,能量梯度和微分耦合。它已经在配置空间中完全饱和,可以通过轨迹引导点采样方法覆盖所有可能的反应路径。此H d不仅可以对涉及S 0的甲醛进行光解反应进行准确的全尺寸动态模拟和S 1,也为将自旋-轨道偶合纳入非绝热骨架提供了至关重要的成分,因此最终使人们能够在同一基础上研究甲醛的内部转化和系统间交叉。










































 京公网安备 11010802027423号
京公网安备 11010802027423号