当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Gas-Phase Binding Energies and Dissociation Dynamics of 1-Alkyl-3-Methylimidazolium Tetrafluoroborate Ionic Liquid Clusters
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-24 , DOI: 10.1021/acs.jpca.0c06297 H. A. Roy 1 , L. A. Hamlow 1 , M. T. Rodgers 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-24 , DOI: 10.1021/acs.jpca.0c06297 H. A. Roy 1 , L. A. Hamlow 1 , M. T. Rodgers 1
Affiliation
|
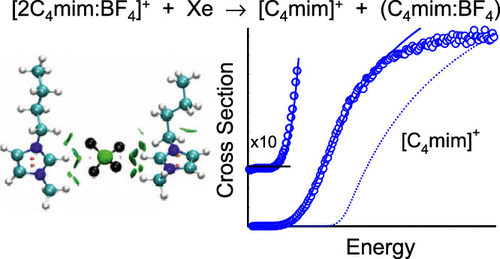
Ionic liquids (ILs) have become increasingly popular due to their useful and unique properties, yet there are still many unanswered questions regarding their fundamental interactions. In particular, details regarding the nature and strength of the intrinsic cation–anion interactions and how they influence the macroscopic properties of ILs are still largely unknown. Elucidating the molecular-level details of these interactions is essential to the development of better models for describing ILs and enabling the purposeful design of ILs with properties tailored for specific applications. Current uses of ILs are widespread and diverse and include applications for energy storage, electrochemistry, designer/green solvents, separations, and space propulsion. To advance the understanding of the energetics, conformations, and dynamics of gas-phase IL clustering relevant to space propulsion, threshold collision-induced dissociation approaches are used to measure the bond dissociation energies (BDEs) of the 2:1 clusters of 1-alkyl-3-methylimidazolium cations and tetrafluoroborate, [2Cnmim:BF4]+. The cation, [Cnmim]+, is varied across the series, 1-ethyl-3-methylimidazolium [C2mim]+, 1-butyl-3-methylimidazolium [C4mim]+, 1-hexyl-3-methylimidazolium [C6mim]+, and 1-octyl-3-methylimidazolium [C8mim]+, to examine the structural and energetic effects of the size of the 1-alkyl substituent on binding. Complementary electronic structure calculations are performed to determine the structures and energetics of the [Cnmim]+ and [BF4]− ions and their binding preferences in the (Cnmim:BF4) ion pairs and [2Cnmim:BF4]+ clusters. Several levels of theory, B3LYP, B3LYP-GD3BJ, and M06-2X, using the 6-311+G(d,p) basis set for geometry optimizations and frequency analyses and the 6-311+G(2d,2p) basis set for energetics, are benchmarked to examine their abilities to properly describe the nature of the binding interactions and to reproduce the measured BDEs. The modest structural variation among these [Cnmim]+ cations produces only minor structural changes and variation in the measured BDEs of the [2Cnmim:BF4]+ clusters. Present findings indicate that the dominant cation–anion interactions involve the 3-methylimidazolium moieties and that these clusters are sufficiently small that differences in packing effects associated with the variable length of the 1-alkyl substituents are not yet significant.
中文翻译:

1-烷基-3-甲基咪唑鎓四氟硼酸根离子液体团簇的气相结合能和解离动力学
离子液体(ILs)由于其有用和独特的特性而变得越来越流行,但是关于它们的基本相互作用仍然存在许多未解决的问题。尤其是,关于内在阳离子与阴离子相互作用的性质和强度以及它们如何影响离子液体宏观性质的细节仍然未知。阐明这些相互作用的分子水平细节对于开发用于描述IL的更好模型并实现针对特定应用量身定制的具有特性的IL的有目的设计至关重要。IL的当前用途广泛且多样,包括能量存储,电化学,设计/绿色溶剂,分离和空间推进的应用。为了加深对能量学,构象的理解,n mim:BF 4 ] +。阳离子[C n mim] +在整个系列中有所不同,1-乙基-3-甲基咪唑鎓[C 2 mim] +,1-丁基-3-甲基咪唑鎓[C 4 mim] +,1-己基-3-甲基咪唑鎓[C 6 mim] +和1-辛基-3-甲基咪唑鎓[C 8 mim] +,以研究1-烷基取代基大小对结合的结构和能量影响。进行互补的电子结构计算以确定[C n mim] +和[BF 4 ]的结构和能量] -离子及其在(C n mim:BF 4)离子对和[2C n mim:BF 4 ] +簇中的结合偏好。几个级别的理论,B3LYP,B3LYP-GD3BJ和M06-2X,使用6-311 + G(d,p)基础集进行几何优化和频率分析,并使用6-311 + G(2d,2p)基础集对于能量学,要进行基准测试,以检查其正确描述结合相互作用的性质并重现所测BDE的能力。这些[C n mim] +阳离子之间的适度结构变化仅产生较小的结构变化,并且[2C n mim:BF 4 ]的BDE实测值也发生变化。+集群。目前的发现表明,主要的阳离子-阴离子相互作用涉及3-甲基咪唑鎓部分,并且这些簇足够小,以致与1-烷基取代基的可变长度相关的堆积效应的差异尚不明显。
更新日期:2020-12-10
中文翻译:
1-烷基-3-甲基咪唑鎓四氟硼酸根离子液体团簇的气相结合能和解离动力学
离子液体(ILs)由于其有用和独特的特性而变得越来越流行,但是关于它们的基本相互作用仍然存在许多未解决的问题。尤其是,关于内在阳离子与阴离子相互作用的性质和强度以及它们如何影响离子液体宏观性质的细节仍然未知。阐明这些相互作用的分子水平细节对于开发用于描述IL的更好模型并实现针对特定应用量身定制的具有特性的IL的有目的设计至关重要。IL的当前用途广泛且多样,包括能量存储,电化学,设计/绿色溶剂,分离和空间推进的应用。为了加深对能量学,构象的理解,n mim:BF 4 ] +。阳离子[C n mim] +在整个系列中有所不同,1-乙基-3-甲基咪唑鎓[C 2 mim] +,1-丁基-3-甲基咪唑鎓[C 4 mim] +,1-己基-3-甲基咪唑鎓[C 6 mim] +和1-辛基-3-甲基咪唑鎓[C 8 mim] +,以研究1-烷基取代基大小对结合的结构和能量影响。进行互补的电子结构计算以确定[C n mim] +和[BF 4 ]的结构和能量] -离子及其在(C n mim:BF 4)离子对和[2C n mim:BF 4 ] +簇中的结合偏好。几个级别的理论,B3LYP,B3LYP-GD3BJ和M06-2X,使用6-311 + G(d,p)基础集进行几何优化和频率分析,并使用6-311 + G(2d,2p)基础集对于能量学,要进行基准测试,以检查其正确描述结合相互作用的性质并重现所测BDE的能力。这些[C n mim] +阳离子之间的适度结构变化仅产生较小的结构变化,并且[2C n mim:BF 4 ]的BDE实测值也发生变化。+集群。目前的发现表明,主要的阳离子-阴离子相互作用涉及3-甲基咪唑鎓部分,并且这些簇足够小,以致与1-烷基取代基的可变长度相关的堆积效应的差异尚不明显。










































 京公网安备 11010802027423号
京公网安备 11010802027423号