当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-Principles Calculation of 1H NMR Chemical Shifts of Complex Metal Polyhydrides: The Essential Inclusion of Relativity and Dynamics
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.inorgchem.0c02753 Abril C Castro 1 , David Balcells 1 , Michal Repisky 2 , Trygve Helgaker 1 , Michele Cascella 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.inorgchem.0c02753 Abril C Castro 1 , David Balcells 1 , Michal Repisky 2 , Trygve Helgaker 1 , Michele Cascella 1
Affiliation
|
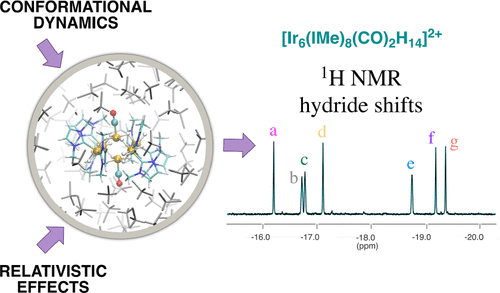
1H NMR spectroscopy has become an important technique for the characterization of transition-metal hydride complexes, whose metal-bound hydrides are often difficult to locate by X-ray diffraction. In this regard, the accurate prediction of 1H NMR chemical shifts provides a useful, but challenging, strategy to help in the interpretation of the experimental spectra. In this work, we establish a density-functional-theory protocol that includes relativistic, solvent, and dynamic effects at a high level of theory, allowing us to report an accurate and reliable interpretation of 1H NMR hydride chemical shifts of iridium polyhydride complexes. In particular, we have studied in detail the hydride chemical shifts of the [Ir6(IMe)8(CO)2H14]2+ complex in order to validate previous assignments. The computed 1H NMR chemical shifts are strongly dependent on the relativistic treatment, the choice of the DFT exchange–correlation functional, and the conformational dynamics. By combining a fully relativistic four-component electronic-structure treatment with ab initio molecular dynamics, we were able to reliably model both the terminal and bridging hydride chemical shifts and to show that two NMR hydride signals were inversely assigned in the experiment.
中文翻译:

复杂金属多氢化物的 1H NMR 化学位移的第一性原理计算:相对论和动力学的本质包容
1 H NMR 光谱已成为表征过渡金属氢化物配合物的重要技术,其金属结合氢化物通常难以通过 X 射线衍射定位。在这方面, 1 H NMR 化学位移的准确预测提供了一种有用但具有挑战性的策略来帮助解释实验光谱。在这项工作中,我们建立了一个密度泛函理论协议,其中包括高水平理论的相对论、溶剂和动态效应,使我们能够准确可靠地解释聚氢化铱络合物的1 H NMR 氢化物化学位移。特别是,我们详细研究了[Ir 6 (IMe) 8 (CO) 2 H 14 ] 2+络合物的氢化物化学位移,以验证先前的分配。计算的1 H NMR 化学位移强烈依赖于相对论处理、DFT 交换相关函数的选择以及构象动力学。通过将完全相对论四分量电子结构处理与从头算分子动力学相结合,我们能够可靠地模拟末端和桥接氢化物化学位移,并表明两个 NMR 氢化物信号在实验中反向分配。
更新日期:2020-12-07
中文翻译:
复杂金属多氢化物的 1H NMR 化学位移的第一性原理计算:相对论和动力学的本质包容
1 H NMR 光谱已成为表征过渡金属氢化物配合物的重要技术,其金属结合氢化物通常难以通过 X 射线衍射定位。在这方面, 1 H NMR 化学位移的准确预测提供了一种有用但具有挑战性的策略来帮助解释实验光谱。在这项工作中,我们建立了一个密度泛函理论协议,其中包括高水平理论的相对论、溶剂和动态效应,使我们能够准确可靠地解释聚氢化铱络合物的1 H NMR 氢化物化学位移。特别是,我们详细研究了[Ir 6 (IMe) 8 (CO) 2 H 14 ] 2+络合物的氢化物化学位移,以验证先前的分配。计算的1 H NMR 化学位移强烈依赖于相对论处理、DFT 交换相关函数的选择以及构象动力学。通过将完全相对论四分量电子结构处理与从头算分子动力学相结合,我们能够可靠地模拟末端和桥接氢化物化学位移,并表明两个 NMR 氢化物信号在实验中反向分配。










































 京公网安备 11010802027423号
京公网安备 11010802027423号