当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multireference Description of Nickel–Aryl Homolytic Bond Dissociation Processes in Photoredox Catalysis
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.jpca.0c08646 David A. Cagan 1 , Gautam D. Stroscio 1 , Alexander Q. Cusumano 1 , Ryan G. Hadt 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.jpca.0c08646 David A. Cagan 1 , Gautam D. Stroscio 1 , Alexander Q. Cusumano 1 , Ryan G. Hadt 1
Affiliation
|
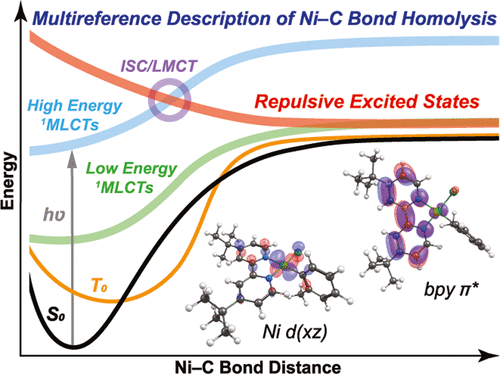
Multireference electronic structure calculations consistent with known experimental data have elucidated a novel mechanism for photo-triggered Ni(II)–C homolytic bond dissociation in Ni 2,2′-bipyridine (bpy) photoredox catalysts. Previously, a thermally assisted dissociation from the lowest energy triplet ligand field excited state was proposed and supported by density functional theory (DFT) calculations that reveal a barrier of ∼30 kcal mol–1. In contrast, multireference ab initio calculations suggest that this process is disfavored, with barrier heights of ∼70 kcal mol–1, and highlight important ligand noninnocent and multiconfigurational contributions to excited state relaxation and bond dissociation processes that are not captured with DFT. In the multireference description, photo-triggered Ni(II)–C homolytic bond dissociation occurs via initial population of a singlet Ni(II)-to-bpy metal-to-ligand charge transfer (1MLCT) excited state, followed by intersystem crossing and aryl-to-Ni(III) charge transfer, overall a formal two-electron transfer process driven by a single photon. This results in repulsive triplet excited states from which spontaneous homolytic bond dissociation can occur, effectively competing with relaxation to the lowest energy nondissociative triplet Ni(II) ligand field excited state. These findings guide important electronic structure considerations for the experimental and computational elucidation of the mechanisms of ground and excited state cross-coupling catalysis mediated by Ni heteroaromatic complexes.
中文翻译:

镍-芳基同质键解离过程在光氧化还原催化中的多参考描述
与已知实验数据一致的多参考电子结构计算阐明了Ni 2,2′-联吡啶(bpy)光氧化还原催化剂中光引发的Ni(II)-C均价键解离的新机理。以前,有人提出了从最低能量三重态配体场激发态进行热辅助解离的方法,并得到密度泛函理论(DFT)计算的支持,该计算揭示了约30 kcal mol –1的势垒。相比之下,多参考从头算则表明该过程不利,势垒高度约为70 kcal mol –1,并突出了重要的配体非纯和多构型对DFT未捕获的激发态弛豫和键解离过程的贡献。在多参考文献的描述中,光触发的Ni(II)-C均价键解离通过单峰Ni(II)向bpy金属向配体的电荷转移的初始填充而发生(1MLCT)激发态,然后进行系统间交叉和芳基到Ni(III)的电荷转移,总体上是由单个光子驱动的正式的两电子转移过程。这导致排斥的三重态激发态,从中可以发生自发的均裂键解离,有效地与弛豫竞争到最低能量的非解离的三重态Ni(II)配体场激发态。这些发现指导重要的电子结构考虑因素,用于实验和计算阐明由Ni杂芳族络合物介导的基态和激发态交叉偶联催化的机理。
更新日期:2020-12-03
中文翻译:
镍-芳基同质键解离过程在光氧化还原催化中的多参考描述
与已知实验数据一致的多参考电子结构计算阐明了Ni 2,2′-联吡啶(bpy)光氧化还原催化剂中光引发的Ni(II)-C均价键解离的新机理。以前,有人提出了从最低能量三重态配体场激发态进行热辅助解离的方法,并得到密度泛函理论(DFT)计算的支持,该计算揭示了约30 kcal mol –1的势垒。相比之下,多参考从头算则表明该过程不利,势垒高度约为70 kcal mol –1,并突出了重要的配体非纯和多构型对DFT未捕获的激发态弛豫和键解离过程的贡献。在多参考文献的描述中,光触发的Ni(II)-C均价键解离通过单峰Ni(II)向bpy金属向配体的电荷转移的初始填充而发生(1MLCT)激发态,然后进行系统间交叉和芳基到Ni(III)的电荷转移,总体上是由单个光子驱动的正式的两电子转移过程。这导致排斥的三重态激发态,从中可以发生自发的均裂键解离,有效地与弛豫竞争到最低能量的非解离的三重态Ni(II)配体场激发态。这些发现指导重要的电子结构考虑因素,用于实验和计算阐明由Ni杂芳族络合物介导的基态和激发态交叉偶联催化的机理。










































 京公网安备 11010802027423号
京公网安备 11010802027423号