当前位置:
X-MOL 学术
›
Macromol. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Examining the Spin State and Redox Chemistry of Ni(Diimine) Catalysts during the Synthesis of π‐Conjugated Polymers
Macromolecular Chemistry and Physics ( IF 2.5 ) Pub Date : 2020-11-23 , DOI: 10.1002/macp.202000321 Adam A. Pollit 1 , Alan J. Lough 1 , Dwight S. Seferos 1, 2
Macromolecular Chemistry and Physics ( IF 2.5 ) Pub Date : 2020-11-23 , DOI: 10.1002/macp.202000321 Adam A. Pollit 1 , Alan J. Lough 1 , Dwight S. Seferos 1, 2
Affiliation
|
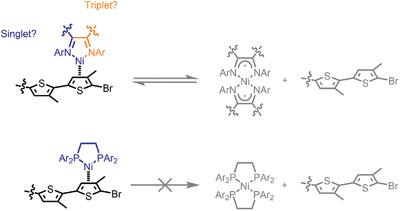
Kumada catalyst transfer polymerization (KCTP) is currently an unparalleled catalytic method for preparing π‐conjugated materials with well‐defined end‐groups, targeted molecular weights, and narrow dispersity. Polymerization control is both catalyst and monomer dependent. Polymerizations using bidentate Ni(phosphine) catalysts and 3‐alkylthiophene (or related) monomers lead to the highest degrees of polymerization control; however the scope of monomers that Ni(phosphines) can polymerize is quite limited. Here, two possible mechanistic limitations of Ni(diimine) catalysts are evaluated: 1) the role of spin states, and 2) the redox non‐innocence of diimine ligands. Density functional theory (DFT) calculations are utilized to evaluate singlet and triplet spin states of Ni(diimine) species throughout the KCTP catalytic cycle. It is found that in the energetically favored triplet state, Ni(diimine) catalyst dissociation from polymer chains is competitive with intramolecular oxidative addition, which reduces polymerization control. The redox activity of Ni(diimine) catalysts during polymerization is investigated and it is determined that a Ni(diimine)X2 precatalyst is converted into Ni(diimine)2 under reductive conditions analogous to KCTP, but surprisingly this species is also active during polymerization which leads to an interesting off‐cycle pathway that has not been previously considered. These insights differentiate the mechanisms of Ni(diimine) and Ni(phosphine) polymerizations, highlighting catalyst spin and redox activity as additional factors in KCTP.
中文翻译:

在π共轭聚合物合成过程中研究Ni(二胺)催化剂的自旋态和氧化还原化学
熊田催化剂转移聚合(KCTP)目前是制备具有明确端基,目标分子量和窄分散性的π共轭材料的无与伦比的催化方法。聚合控制既取决于催化剂又取决于单体。使用双齿Ni(膦)催化剂和3-烷基噻吩(或相关)单体进行聚合可实现最高的聚合控制程度;但是,Ni(膦)可以聚合的单体范围非常有限。在此,评估了Ni(diimine)催化剂的两个可能的机理局限性:1)自旋态的作用,以及2)diimine配体的氧化还原无毒。密度泛函理论(DFT)计算用于评估整个KCTP催化循环中Ni(diimine)物种的单重态和三重态自旋态。发现在能量有利的三重态下,从聚合物链解离的Ni(二亚胺)催化剂与分子内氧化加成竞争,这降低了聚合控制。研究了Ni(diimine)催化剂在聚合过程中的氧化还原活性,并确定了Ni(diimine)X2预催化剂在类似于KCTP的还原条件下转化为Ni(diimine )2,但令人惊讶的是,该物种在聚合过程中也具有活性,这导致了以前没有考虑过的有趣的循环外途径。这些见解区分了Ni(diimine)和Ni(phosphine)聚合的机理,强调了催化剂自旋和氧化还原活性是KCTP中的其他因素。
更新日期:2020-12-18
中文翻译:
在π共轭聚合物合成过程中研究Ni(二胺)催化剂的自旋态和氧化还原化学
熊田催化剂转移聚合(KCTP)目前是制备具有明确端基,目标分子量和窄分散性的π共轭材料的无与伦比的催化方法。聚合控制既取决于催化剂又取决于单体。使用双齿Ni(膦)催化剂和3-烷基噻吩(或相关)单体进行聚合可实现最高的聚合控制程度;但是,Ni(膦)可以聚合的单体范围非常有限。在此,评估了Ni(diimine)催化剂的两个可能的机理局限性:1)自旋态的作用,以及2)diimine配体的氧化还原无毒。密度泛函理论(DFT)计算用于评估整个KCTP催化循环中Ni(diimine)物种的单重态和三重态自旋态。发现在能量有利的三重态下,从聚合物链解离的Ni(二亚胺)催化剂与分子内氧化加成竞争,这降低了聚合控制。研究了Ni(diimine)催化剂在聚合过程中的氧化还原活性,并确定了Ni(diimine)X2预催化剂在类似于KCTP的还原条件下转化为Ni(diimine )2,但令人惊讶的是,该物种在聚合过程中也具有活性,这导致了以前没有考虑过的有趣的循环外途径。这些见解区分了Ni(diimine)和Ni(phosphine)聚合的机理,强调了催化剂自旋和氧化还原活性是KCTP中的其他因素。










































 京公网安备 11010802027423号
京公网安备 11010802027423号